
Abstract
Background:
Walking impairment causes disability and reduced quality of life in patients with multiple sclerosis (MS).
Objective:
Characterize the safety and efficacy of ADS-5102 (amantadine) extended release capsules, 274 mg administered once daily at bedtime in patients with MS with walking impairment.
Methods:
This randomized, double-blind, placebo-controlled, 4-week study was conducted at 14 trial sites in the United States. Study objectives included safety and tolerability of ADS-5102, and efficacy assessments (Timed 25-Foot Walk (T25FW), Timed Up and Go (TUG), 2-Minute Walk Test, and Multiple Sclerosis Walking Scale-12). Fatigue, depression, and cognition also were assessed.
Results:
A total of 60 patients were randomized (30 to ADS-5102 and 30 to placebo); 59 of whom were treated. The most frequent adverse events (AEs) were dry mouth, constipation, and insomnia. Five ADS-5102 patients and no placebo patients discontinued treatment due to AEs. One patient in the ADS-5102 group experienced a serious AE—suspected serotonin syndrome. A 16.6% placebo-adjusted improvement was seen in the T25FW test (p < 0.05). A 10% placebo-adjusted improvement in TUG was also observed. No changes in fatigue, depression, or cognition were observed.
Conclusion:
ADS-5102 was generally well tolerated. These data demonstrate an effect of ADS-5102 on walking speed. Further studies are warranted to confirm these observations.
Introduction
Multiple sclerosis (MS) is a chronic immune-mediated disease of the central nervous system (CNS), with a global prevalence estimated to be 2.3 million people in 2013. 1 In North America and Europe, MS is the leading non-traumatic cause of neurologic disability in young adults. 2 Impaired gait and mobility develops in approximately 75% of people with MS 3 and is a frequently disabling symptom. 4
The potassium channel blocker, fampridine, currently is the only medication approved to treat gait disorders in MS.5,6 Approximately, only 40% of people treated with fampridine are considered responders, based on consistent improvement in walking speed.5,6 Thus, additional medications that improve walking ability and other symptoms of MS are needed.
Animal models suggest that immediate-release amantadine hydrochloride might be beneficial for the treatment of MS. 7 Amantadine hydrochloride has also been used off-label to treat MS-associated fatigue, although the mechanism of action is still unclear.8–14 Immediate-release amantadine hydrochloride is currently indicated for the prophylaxis and treatment of infection caused by influenza A virus 15 and is also approved to treat parkinsonism. Most patients with Parkinson’s disease (PD) tolerate immediate-release amantadine doses of 81–161 mg daily (equivalent to 100–200 mg amantadine hydrochloride), given in divided doses to avoid adverse events (AEs). 15 Higher doses may be more efficacious but are associated with increased CNS AEs. 16
ADS-5102 (amantadine) extended release capsules (GOCOVRITM, Adamas Pharmaceuticals, Inc.) is approved for the treatment of dyskinesia in patients with PD receiving levodopa-based therapy, with or without concomitant dopaminergic medications. 17 The formulation is designed to be administered at bedtime, with a concentration–time curve characterized by a slow initial rise in amantadine concentrations at night with a plateau of high concentrations achieved in the morning and sustained during waking hours. This strategy allows for high plasma concentrations (1500 ng/mL) during daytime that cannot be achieved with immediate-release amantadine. 18 In clinical trials of ADS-5102 to treat PD patients with dyskinesia,18–20 the most common adverse drug reactions, occurring in >10% of ADS-5102–treated patients, were hallucinations, dizziness, dry mouth, peripheral edema, constipation, falls, and orthostatic hypotension. Hallucinations were the most commonly observed clinically relevant reaction, were mostly mild and reversible, did not require intervention, and most did not lead to drug discontinuation.
The mechanism of action related to walking improvement is unclear. Patch clamp electrophysiology studies in rat brain coronal slices in vitro show that amantadine blocks delayed rectifier potassium currents over a wide concentration range (10–1000 µM) and blocks potassium leak currents at higher concentrations (100–1000 µM). Blockade of neuronal potassium channels may result in increased duration of action potentials, improving conduction across demyelinated axonal internodes. In addition, amantadine is known to be an uncompetitive inhibitor of the N-methyl-D-aspartate (NMDA) receptor, and increased glutamatergic signaling is believed to contribute to the pathophysiology in MS. 21 In an experimental autoimmune encephalomyelitis mouse model of MS, amantadine reduced disease severity, improved walking speed, and reduced neuroinflammation as measured by microglia activation. 22
We undertook this study to assess the safety and tolerability of ADS-5102 in patients with MS. Secondary objectives were to assess the potential benefit of ADS-5102 on walking speed, functional mobility, walking distance, fatigue, depression, and cognition of patients with MS.
Methods
Study design and patients
We conducted a randomized, double-blind, placebo-controlled, phase 2 proof-of-concept clinical trial at 14 medical centers and clinical practices in the United States. Eligible patients were aged 18–70 years, with MS fulfilling the 2010 McDonald Criteria, 23 Expanded Disability Status Scale (EDSS) score ⩽6.5, 24 ability to complete two trials of the Timed 25-Foot Walk (T25FW) in 8–45 seconds each, 25 stable disease therapy regimen for ⩾30 days, and a stable antidepressant dose for ⩾60 days if applicable. Key exclusion criteria, evaluated at screening, were MS relapse or corticosteroid treatment within 30 days; administration of fampridine, methylphenidate, modafinil, armodafinil, or other treatment specifically to improve fatigue or walking ability within 30 days; new or changes to physical therapy regimen within 30 days; administration of botulinum toxin to treat spasticity within 3 months; hallucinations within 2 years; seizure within 2 years; intolerance of amantadine; current treatment with medications that may affect the renal clearance of amantadine; administration of medications that prolong the QT interval and have a known risk of torsades de pointes; and an estimated glomerular filtration rate <50 mL/min/1.73 m2.
Central and local ethics committees approved the study. Patients gave written informed consent before any study-related procedures were done. The study was conducted in accordance with the International Conference on Harmonization Guidelines for Good Clinical Practice and the principles of the Declaration of Helsinki. The sponsor designed the study with input from the authors and monitored its conduct. Data were obtained by the investigators and analyzed by the sponsor. This trial is registered with ClinicalTrials.gov, number NCT02471222.
Randomization and masking
Patients who fulfilled eligibility criteria during a maximum of 21 days of screening were randomly assigned with equal probability to ADS-5102 274 mg (equivalent to 340 mg amantadine hydrochloride) or matching placebo. The randomization list was generated and validated by Agility Clinical, Inc., (Carlsbad, CA, USA). Randomization was accomplished through an interactive web-based response system managed by Endpoint Clinical (San Francisco, CA, USA), which allowed for unblinding of individual patient data if necessary for patient safety.
During the trial, patients, study site personnel, and the sponsor study team, including study statistician, were unaware of study group assignments. At the study sites, a treating neurologist supervised medical management, and trained assessor(s) administered the outcome measures.
Procedures
Patients who were randomized to ADS-5102 received 137 mg during Week 1, then 274 mg during Weeks 2–4 in a blinded fashion. Patients were instructed to take the assigned study treatment orally once daily at bedtime for 4 weeks. Study medication or matching placebo was supplied in blister packs which were returned at each visit; trial staff used these to assess treatment compliance.
Safety assessments included monitoring of AEs and vital signs at screening, baseline, Week 2, Week 4, and safety follow-up visit; general physical examination at screening and Week 4; electrocardiogram at screening; complete blood count, serum chemistry, and urinalysis at screening and Week 4; and pregnancy test (if applicable) at screening, baseline, and safety follow-up.
The EDSS 24 was assessed at screening, and the T25FW 25 was assessed at screening, baseline, and Weeks 2 and 4. Other efficacy measures were assessed at baseline, and Weeks 2 and 4, including Timed Up and Go (TUG), 26 2-Minute Walk Test (2MWT), 27 Multiple Sclerosis Walking Scale-12 (MSWS-12). 28 Fatigue was assessed using the Fatigue Scale for Motor and Cognitive Functions (FSMC), 29 depression was assessed using Beck’s Depression Inventory-2 (BDI-2), 30 and cognitive impairment was measured by the Brief International Cognitive Assessment for MS (BICAMS). 31
Outcomes
The primary objective was to evaluate the safety and tolerability of ADS-5102 in patients with MS. The principal secondary outcome measures included change from baseline to Weeks 2 and 4 in walking ability and mobility measured by T25FW, TUG, 2MWT, and MSWS-12. Other secondary outcome measures were change from baseline in FSMC, BDI-2, and BICAMS.
Statistical analysis
The safety analysis population included all randomized patients who received at least one dose of study drug. Safety endpoints were summarized by treatment group from the time of first dose and include all available safety data. No formal statistical testing was performed on the safety data. AEs were presented according to the Medical Dictionary for Regulatory Activities (MedDRA® version 17.0 or higher), system organ class, and preferred term, and the most common AEs (⩾2 ADS-5102-treated patients) were tabulated by treatment group. A patient was counted once if he or she reported one or more AEs.
The efficacy analyses utilized a modified intention-to-treat (mITT) analysis set, defined as all randomized patients who received at least one dose of study medication and provided at least one post-baseline walking assessment.
Comparisons between treatment groups in the mean changes from baseline in the efficacy outcome measures were obtained from an analysis of covariance model, with baseline assessment as covariate and treatment groups as a class effect. This was done separately for each study week. Comparisons of mean relative (percent) changes from baseline were done in a similar manner. The T25FW was also analyzed by determining the proportion of patients with ⩾20% improvement in walking speed compared with baseline, a change considered clinically meaningful. 25 Missing data were imputed as last-observation-carried forward. Values of p <0.05 were considered statistically significant. Because of the exploratory nature of the study, no correction for multiple comparisons was performed.
Role of the funding source
The sponsor contributed to study design, conduct, and reporting. The decision to submit for publication was in collaboration with the coauthors. All external authors had full access to all study data and final responsibility for the decision to submit the report for publication.
Results
Study population
Baseline demographic and clinical characteristics were similar across treatment groups (Table 1). Of 60 patients randomized, 29 (97%) in the placebo group (n = 29) and 24 (80%) in the ADS-5102 group completed the study (Figure 1). One patient was randomized to placebo in error but not treated; and five ADS-5102 patients discontinued the study due to AEs and one due to noncompliance with the study drug (Figure 1). The mITT population for the efficacy analysis comprised 56 patients (27 in the ADS-5102 group and 29 in the placebo group). The mean (standard deviation (SD)) treatment compliance for all patients were comparable between the treatment groups (99.1% (2.6) placebo group; 98.1% (3.1) ADS-5102 group).
Baseline demographic and clinical characteristics of patients (mITT population).
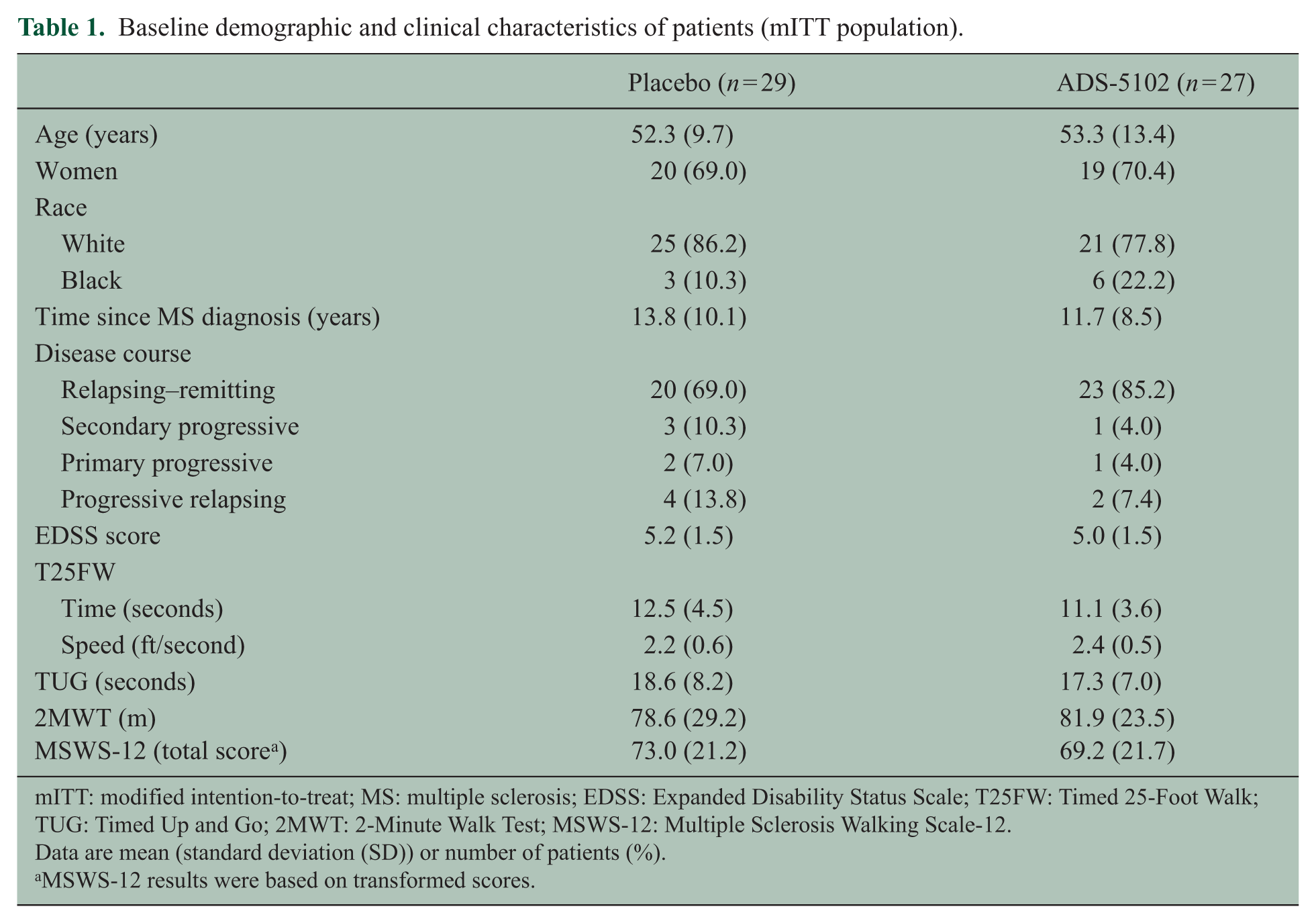
mITT: modified intention-to-treat; MS: multiple sclerosis; EDSS: Expanded Disability Status Scale; T25FW: Timed 25-Foot Walk; TUG: Timed Up and Go; 2MWT: 2-Minute Walk Test; MSWS-12: Multiple Sclerosis Walking Scale-12.
Data are mean (standard deviation (SD)) or number of patients (%).
MSWS-12 results were based on transformed scores.
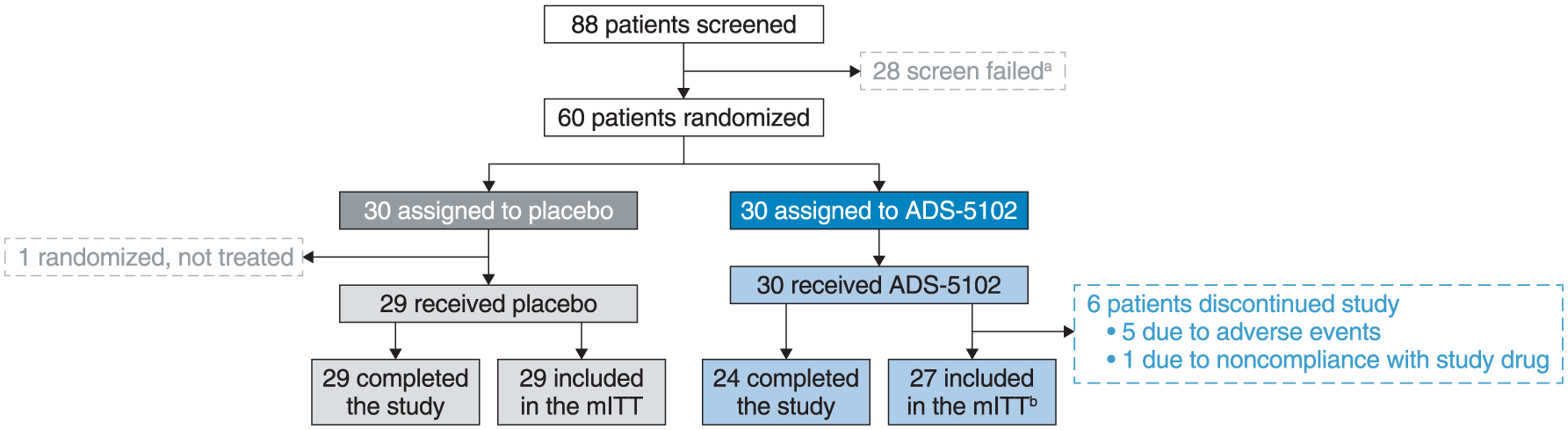
Patient disposition.
Safety and tolerability
Comparable proportions of patients experienced at least one AE, although a higher proportion of ADS-5102-treated patients (30.5%) reported a study drug–related AE compared with the placebo group (20.7%) (Table 2). Most AEs were mild to moderate in severity. The single serious AE in the ADS-5102 group was reported as suspected serotonin syndrome and considered related to treatment. The serious AE occurred 4 days after the last ADS-5102 dose and resolved 74 days after onset. Five patients discontinued study drug due to an AE, all in the ADS-5102 group; the five AEs were duloxetine withdrawal, possible serotonin syndrome, corticosteroid-induced psychosis, dry mouth, and a tooth infection, each in one patient. The most common AEs associated with ADS-5102 were dry mouth (23.3%), constipation (10.0%), and insomnia (10.0%). Other clinically relevant AEs reported in the ADS-5102-treated arms were hallucinations (2, 6.7%), ataxia (2, 6.7%), and agitation (2, 6.7%). No clinically significant abnormalities of vital signs, physical examination, or laboratory values occurred in either treatment group.
AEs (safety population).
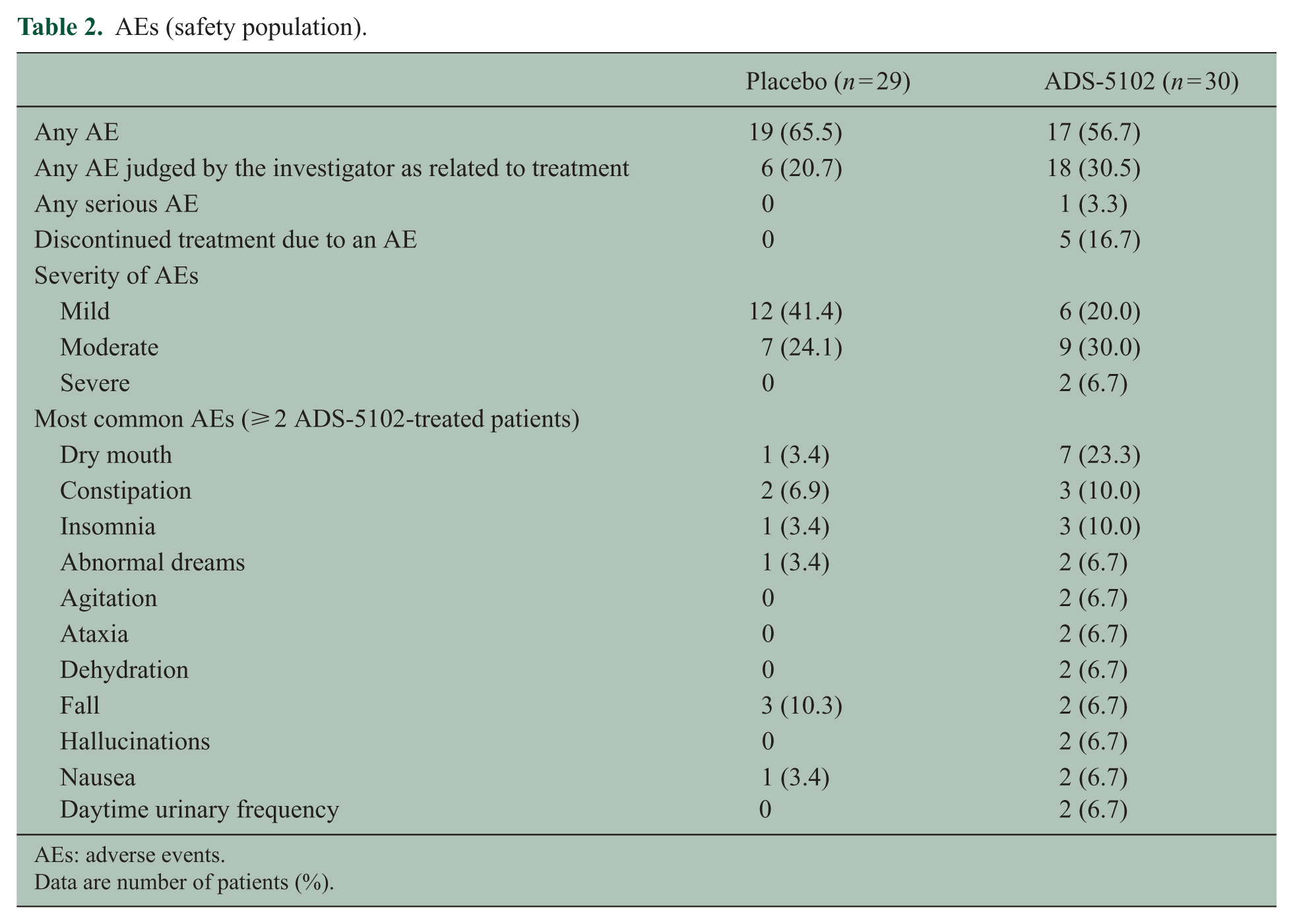
AEs: adverse events.
Data are number of patients (%).
Efficacy
Mean percent improvement compared with baseline in walking speed measured by the T25FW was modest at Week 2 in the placebo group (7.8%) without further improvement at Week 4 (8.0%) (Figure 2(a), Table 3). Improvement in walking speed was greater in the ADS-5102-treated patients at Week 2 (17.1%) and Week 4 (24.6%), with a difference versus placebo at Week 4 of 16.6% (p = 0.04). A greater proportion of ADS-5102-treated patients had a clinically significant (⩾20%) improvement in walking speed compared with the placebo group (30% vs 17%), based on the percent change from baseline in the average of Weeks 2 and 4 (Supplementary Figure e-1). Results in terms of absolute change were consistent with these findings.
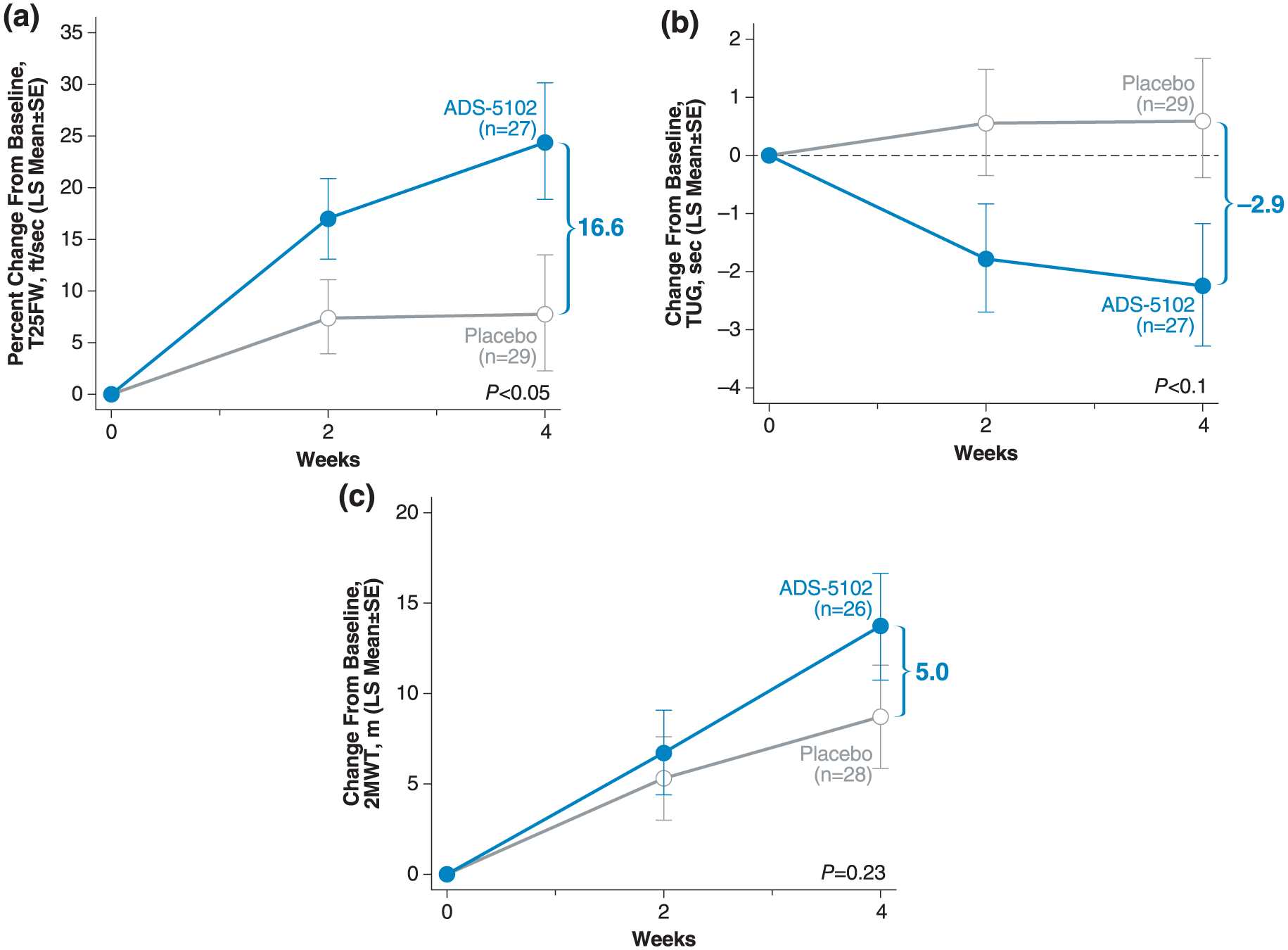
Improvement in: (a) T25FW, (b) TUG, and (c) 2MWT over 4 weeks.
Walking outcome measures: absolute and relative (%) least-squares mean changes from baseline to Week 4 (mITT).
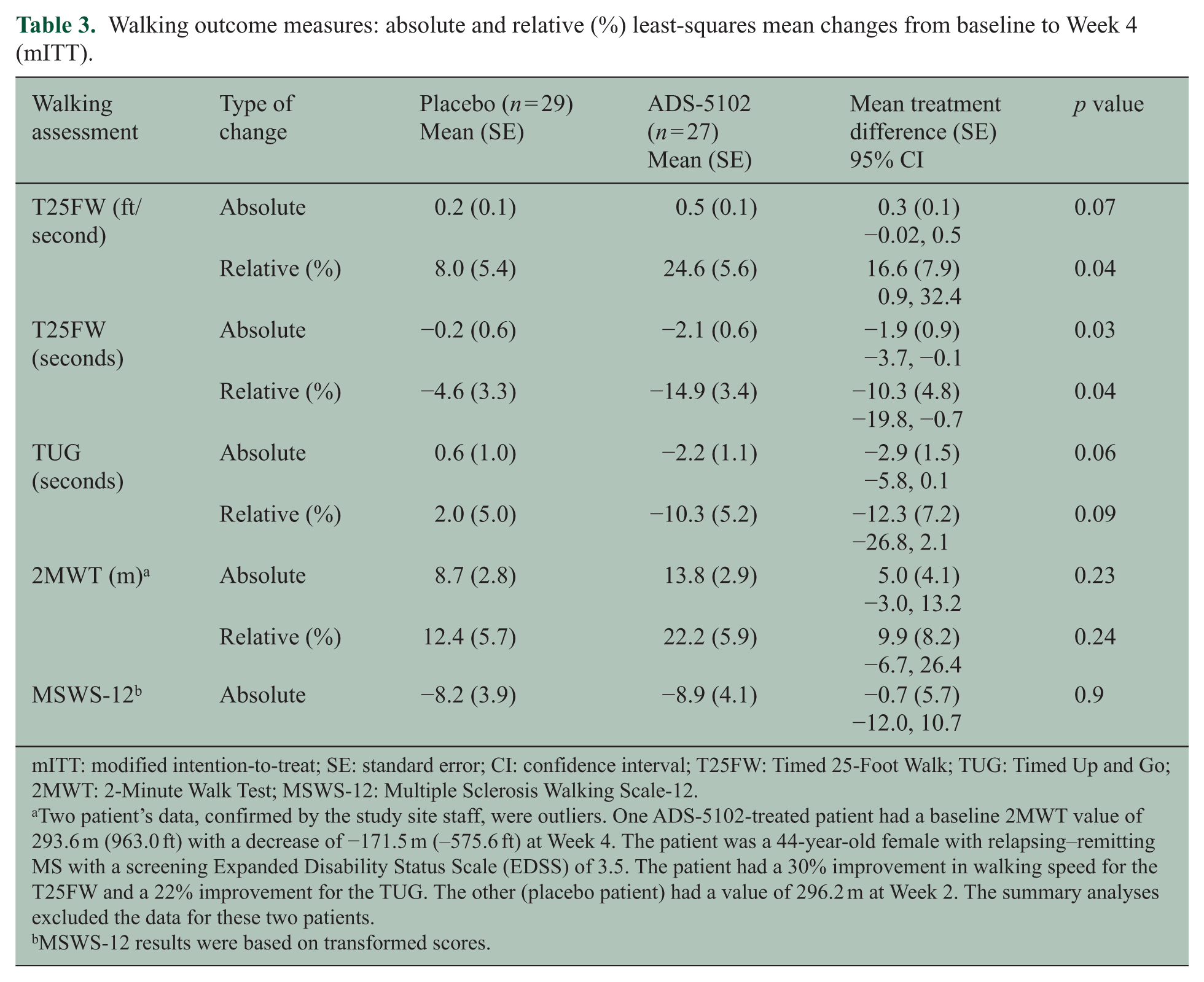
mITT: modified intention-to-treat; SE: standard error; CI: confidence interval; T25FW: Timed 25-Foot Walk; TUG: Timed Up and Go; 2MWT: 2-Minute Walk Test; MSWS-12: Multiple Sclerosis Walking Scale-12.
Two patient’s data, confirmed by the study site staff, were outliers. One ADS-5102-treated patient had a baseline 2MWT value of 293.6 m (963.0 ft) with a decrease of −171.5 m (–575.6 ft) at Week 4. The patient was a 44-year-old female with relapsing–remitting MS with a screening Expanded Disability Status Scale (EDSS) of 3.5. The patient had a 30% improvement in walking speed for the T25FW and a 22% improvement for the TUG. The other (placebo patient) had a value of 296.2 m at Week 2. The summary analyses excluded the data for these two patients.
MSWS-12 results were based on transformed scores.
There were trends for greater improvement in the TUG and 2MWT at Weeks 2 and 4 in the ADS-5102 group compared with the placebo group, although the differences were not significant (Figure 2(b), 2(c)). Results were consistent between the absolute and relative changes. No evidence of an effect of ADS-5102 as compared with placebo was noted at Week 4 for the change from baseline in MSWS-12. Similarly, no statistically significant between-group differences in FSMC, BDI-2, and BICAMS were seen at 2 or 4 weeks.
Discussion
This 4-week, placebo-controlled, proof-of-concept study showed a manageable safety and tolerability profile for ADS-5102 in patients with MS. The safety data were similar to the previously published results in PD.18–20 The types of reported AEs were also consistent with the known safety profile of immediate-release amantadine.
A statistically significant effect on the percent change in walking speed from baseline was observed, and a greater proportion of ADS-5102-treated patients experienced a ⩾20% improvement. Trends suggesting benefit, which did not reach statistical significance, were observed for the TUG, and less so on the 2MWT. Previous studies indicate these tests are less sensitive, requiring longer follow-up and larger sample size to demonstrate benefit. 32 Results on patient-reported walking ability measured by the MSWS-12 over 4 weeks did not demonstrate benefit. The short duration of the study may not have provided sufficient time to detect a treatment effect on this patient-reported outcome since the recall period was 2 weeks. Despite showing benefits to various aspects of walking ability, this study did not identify improvement in fatigue, depression, or cognitive function, possibly due to the short study duration, small sample size, and only mild abnormality at baseline.
While this study provides preliminary evidence that ADS-5102 may improve walking in people with MS and gait impairment, the mechanism of such an effect remains unknown. Potential limitations of this study include a small sample size, short study duration, and lack of formal assessment of blinding during exit interviews. Despite the inclusion of several functional assessments, the study population was only enriched for impaired walking speed (as measured by the T25FW) based on inclusion criteria. Larger studies of longer duration are warranted to confirm the efficacy and tolerability of ADS-5102 in MS patients with walking impairment and to identify predictors of patient response.
Footnotes
Acknowledgements
J.A.C. wrote the first draft of the manuscript. L.L. provided statistical and analysis support. J.A.C., S.F.H., T.R.B., M.G., B.W.T., L.L., C.J.S-P., A.E.R., D.N.C., and R.P. reviewed the study data, reviewed and edited the report, and approved the final version.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: J.A.C. reports personal fees for serving as a consultant to Adamas and Celgene, and as co-editor of Multiple Sclerosis Journal—Experimental, Translational, and Clinical. S.F.H. reports serving as a consultant for AbbVie, Bayer, Genentech/Roche, and Sanofi-Genzyme. He also reports multicenter clinical research trial participation for Actelion, Adamas, Genentech/Roche, Osmotica, and Teva. He has received research grant support from Sanofi-Genzyme and has served on speakers’ bureaus for Mallinckrodt, Novartis, Sanofi-Genzyme, and Teva. T.R.B. has served as a consultant and has received speaking honoraria from Acorda Therapeutics. M.G. reports research grants and or speaking fees from Novartis, Biogen, EMD Serono, TEVA, Adamas, Roche, and Sanofi-Genzyme, outside the submitted work. B.W.T. reports nothing to disclose. D.N.C. is a consultant for Adamas Pharmaceuticals, Inc. He reports having received consulting fees, stock options, and owns stock in Adamas Pharmaceuticals. L.L., C.J.S-P, A.E.R., and R.P. are employees of the funder of this study, Adamas Pharmaceuticals, Inc.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Medical writing and editorial support was provided by The Curry Rockefeller Group, LLC, and was funded by Adamas Pharmaceuticals, Inc. Preliminary results were presented at the ACTRIMS 2017 Forum, 23–25 February 2017, in Orlando, FL, USA, and the Consortium of MS Centers annual meeting, 24–27 May 2017, in New Orleans, LA, USA. This study was funded by Adamas Pharmaceuticals, Inc.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.