
Abstract
Understanding the characteristics of neuromyelitis optica spectrum disorder (NMOSD) with recurrent short partial transverse myelitis (SPTM), which is very rare, contributes to the differential diagnosis of multiple sclerosis (MS). We present two Chinese aquaporin-4 immunoglobulin G (AQP4-IgG)-seropositive NMOSD cases who had at least twice SPTM during 4 and 6 years of follow-up, respectively. Their SPTMs have been mild and responded well to corticosteroids just like in the case of MS. The findings highlight the need of searching for serum AQP4-IgG (cell-based assay strongly recommended) in patients with recurrent SPTM and suggest that those patients may have a mild acute attack phase and favorable long-term prognosis.
Keywords
Neuromyelitis optica spectrum disorder (NMOSD) is an inflammatory central nervous system (CNS) disorder distinct from multiple sclerosis (MS) and is associated with serum aquaporin-4 immunoglobulin G (AQP4-IgG) antibodies. 1 The treatment and natural course are substantially different between NMOSD and MS. Consensus guidelines favor oral corticosteroids with a concomitant immunosuppressive agent, which are probably ineffective for MS, to prevent recurrent NMOSD attacks. Disease-modifying treatments for MS are probably ineffective for NMOSD and might even be harmful. 2 As long-term prognosis is concerned, the time to walking aid needed of NMOSD patients is much shorter (7.8 years) than that noted even in untreated relapsing-remitting MS patients (23 years). 3 Moreover, there is a unique form of relapsing demyelinating disease in the Asian population named opticospinal MS, which appears to be an admixture of conventional MS and NMOSD. Thus, the differential diagnosis between NMOSD and MS has always been important and difficult.
There are indeed certain clinical features that help in the differential diagnosis. As myelitis is concerned, a complete (rather than partial) spinal cord syndrome, ⩽3 vertebral segments longitudinally extensive transverse myelitis (LETM) lesions is particularly suggestive of NMOSD. 1 However, it is not pathognomonic. For example, Bourre et al. 4 found 1 out of 85 patients with first-episode acute partial transverse myelitis to have NMO after a mean follow-up of 8.7 years. Recent studies demonstrated that 7%–14.5% of initial and 8% of subsequent myelitis episodes in AQP4-IgG-seropositive patients involved short transverse myelitis (STM, <3 vertebral segments).5–7 These findings suggest that acute partial transverse myelitis is rare, but STM is not uncommon in NMOSD. In terms of recurrent short partial transverse myelitis (SPTM) in NMOSD, there have been very rare reports so far. Herein, we present for the first time two Chinese AQP4-IgG-seropositive NMOSD cases with SPTM.
Methods
NMOSD is defined according to the proposal of Wingerchuk et al. 1 Cases were observed between July 2013 and October 2016 in our own case database. Inclusion criteria were as follows: (1) clinical presentations of all acute myelitis episodes suggested partial transverse lesions, (2) magnetic resonance imaging (MRI) performed in the acute attack stage showed spinal cord T2-hyperintense lesions <3 vertebral segments, (3) AQP4-IgG seropositivity, and (4) final diagnosis of NMO or NMOSD. We excluded patients with SPTM who had one or more LETM attack. AQP4-IgG serostatus was evaluated by tissue-based indirect immunofluorescence. All patients signed written informed consent.
A total of 21 NMOSD patients with AQP4-IgG seropositivity were identified from our own case database. Two patients with SPTM were excluded: one with initial episode of SPTM developed subsequent LETM; another with initial episode of LETM developed subsequent five SPTM. At last, two cases with at least two SPTM were determined (2/21, 9.5%; Table 1).
Clinical features of AQP4-IgG-seropositive NMOSD cases with recurrent SPTM.

SPTM: short partial transverse myelitis; EDSS: Expanded Disability Status Scale; ARR: annualized relapse rate; LON: left optic neuritis; RON: right optic neuritis.
Case report
Case 1
A 33-year-old woman initially experienced a sudden onset of right lower limb numbness on 4 May 2012. In the following week, disease progressed gradually with the occurrence of pain and a girdle sensation of the right costal arch, left lower limb numbness, and left optic neuritis. Spine MRI showed two divided T2-hyperintensity lesions with one vertebral segment long in the thoracic cord (Figure 1). Cerebrospinal fluid (CSF) analysis revealed no alterations. Serum AQP4-IgG was positive. In July 2014, the patient experienced a second attack which presented hypoalgesia below the middle calf level with decreased tendon reflexes and negative Babinski signs of both legs, and left optic neuritis peaked within 8 days. The second serum AQP4-IgG test was still positive. Spine MRI showed no definite signal change. Head MRI showed several nonspecific subcortical hyperdense punctuate lesions (Figure 1). In June 2015, the patient experienced right optic neuritis. In October 2016, she developed a fourth attack presented with left nucho-occipital itch and right limb numbness. Spine MRI showed two divided T2-hyperintensity lesions with one vertebral segment long in the cervical cord (Figure 1). During each acute phase, the patient responded very well to a 3-day course of intravenous methylprednisolone (IVMP) 1 g daily. Due to concerns about side effects of azathioprine and prednisolone, she did not insist on taking them for over 1 year between the two attacks.
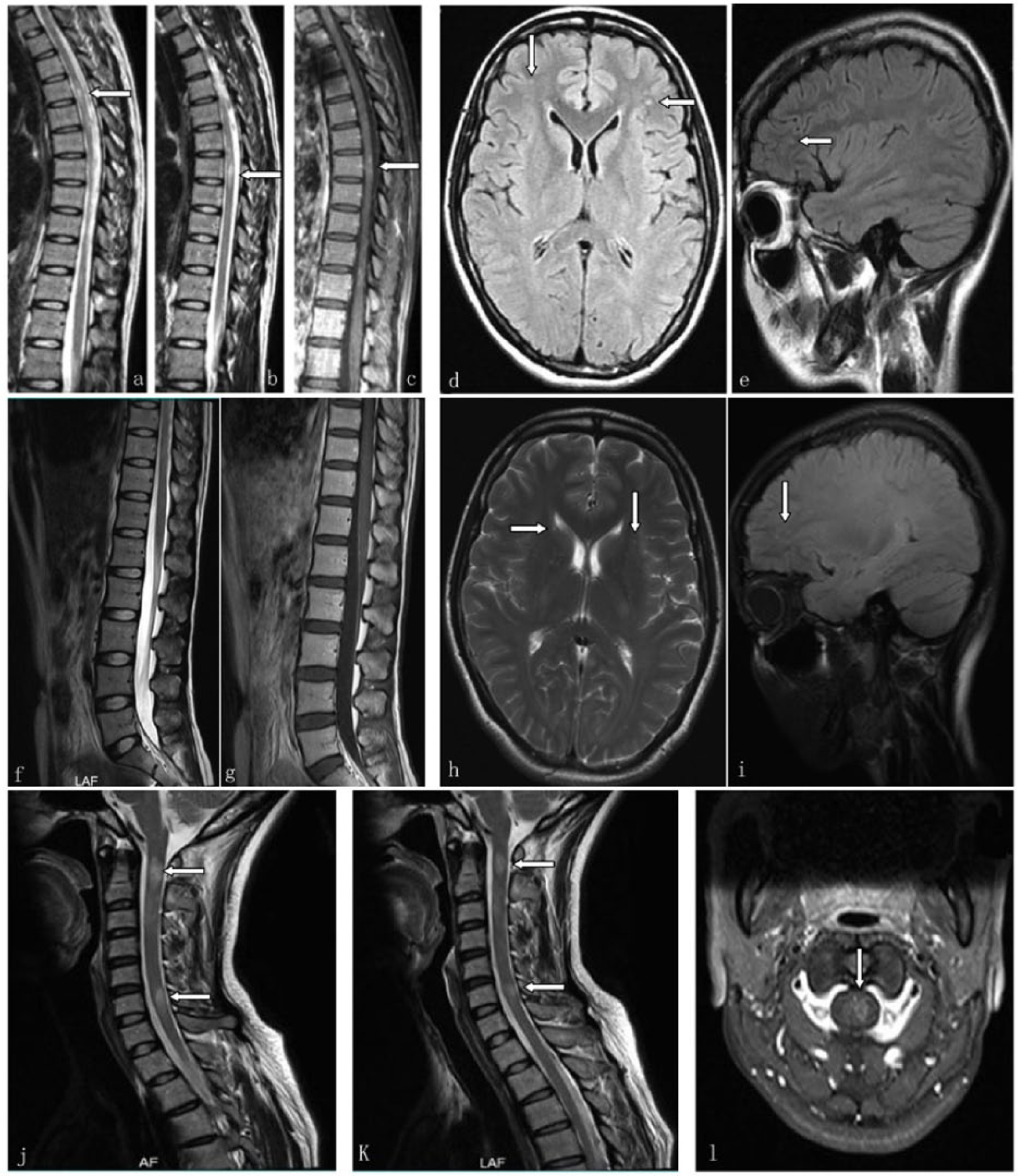
On 4 May 2012, (a and b—arrow) spine MRI exhibited two isolated lesions with one vertebral segment long on the sagittal T2-weighted sequence, (c—arrow) one of which was mildly enhanced after injection of contrast agent. On 5 May 2012, head MRI exhibited nonspecific subcortical punctuate lesions at frontal lobes on (d—arrows) axial and (e—arrow) sagittal FLAIR sequences. On 29 March 2014, MRI of the lumbosacral spinal cord showed no definite signal change on sagittal (f) T2 and (g) T1 contrast sequences. On 29 March 2014, head MRI showed several nonspecific hyperdense punctuate lesions at frontal lobes on (h) axial T2 and (i) sagittal FLAIR sequences. (j and k—arrows) On 10 and 25 October 2016, spine MRI showed two divided lesions with one vertebral segment long in the cervical cord on the sagittal T2-weighted sequence. (l—arrow) Axial T1 contrast sequence showed the lesion located in the left part of cord.
Case 2
A 28-year-old woman had experienced three attacks before the first SPTM, including right optic neuritis once and brainstem syndromes twice (Figure 2) since March 2010. The first SPTM presented left occipital neuralgia and itch in the left nuchal area with a C1–2 lesion demonstrated by spine MRI in June 2012 (Figure 2). CSF analysis revealed no alterations. Serum AQP4-IgG was positive. After a 3-day course of IVMP 1 g daily followed by taking prednisolone and azathioprine (2 mg/kg d) orally for the maintenance treatment, she recovered completely. Due to concerns about side effects, she discontinued the maintenance therapy by herself in July 2013. After 9 months, she developed relapse involving the brainstem. The second serum AQP4-IgG test was still positive. In June 2016, she developed a second SPTM presented with itch and a girdle sensation at breast level with bilateral leg numbness. Spine MRI showed two isolated T2-hyperintensity lesions about one vertebral segment long in the thoracic cord (Figure 2). After a 3-day course of IVMP 1 g daily followed by taking prednisolone and azathioprine (2 mg/kg d) orally for the maintenance treatment, she recovered gradually.
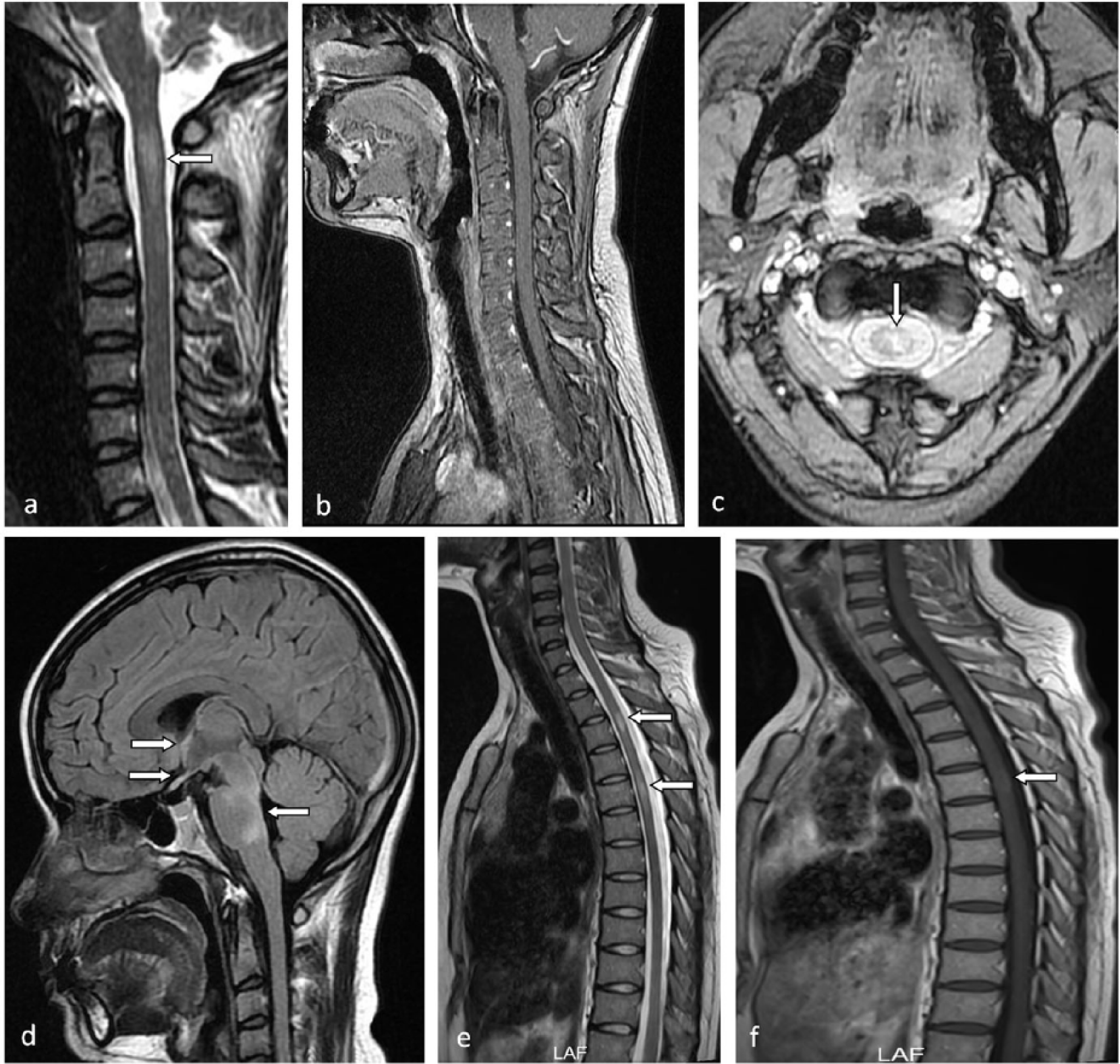
On 6 June 2012, spine MRI exhibited a lesion which was <2 vertebral segments on the (a—arrow) sagittal T2-weighted sequence, (b) without enhancement, in the left half of the cord on the (c—arrow) axial T2-weighted sequence. On 3 August 2011, (d) the sagittal FLAIR of head MRI showed hyperdense lesions located at hypothalamus, optic chiasm, midbrain, and pontine in the acute phase of the third attack. On 8 June 2016, spine MRI showed two isolated lesions <2 vertebral segments located at T3 and T5 vertebral levels on the (e—arrows) sagittal T2-weighted sequence, (f—arrow) one of which had mild enhancement after injection of contrast agent.
Discussion
The present findings suggest that SPTM is not uncommon in a Chinese AQP4-IgG-seropositive NMOSD cohort. It can pose a serious differential diagnostic challenge between NMOSD and MS, if AQP4-IgG evaluation is unavailable. Consistent with the well-known consensus on NMOSD diagnosis, the two cases lack oligoclonal bands (OCB) in the CSF and have no MS-typical lesions on head MRI. 1 This indeed confirmed the differential diagnostic value of OCB in the CSF and brain MRI abnormalities. Previous studies have demonstrated the presence of CSF OCB as supportive evidence for MS and a red flag for NMOSD; the presence of some MRI lesion patterns, including Dawson fingers, periventricular lesions located in the inferior temporal lobe, and cortical lesions, is considered to be typical of MS and a red flag for NMOSD. 1
The onset age of the present cases is similar to that of MS patients (average 29 years),8,9 but lower than that of NMOSD patients (average 39 years). 8 Consistent with previous reports, SPTM of NMOSD responds well to corticosteroids just like MS does in the acute attack phase. 6 In one case, the patient has no disability during 4 years of follow-up, and in another case, the patient has only mild disability in the sensory system during 6 years of follow-up. All of this suggests that NMOSD with recurrent SPTM has many similarities with MS and has less severity as well as better long-term prognosis than typical NMOSD with LETM.
In addition, both cases are susceptible to having two isolated spinal cord lesions in one episode. This suggests that dissemination in space in the spinal cord is also common in NMOSD. Similar to a previous study, itch was the prominent symptom in these two cases. 10 This is consistent with the central location of spinal cord of NMOSD lesions. Presumably, inflammation/demyelination involving second-order itch neurons in the dorsal horn of the spinal cord is responsible for the itch. 10
This study also reflects a reality that patients with recurrent SPTM have poor compliance to immunosuppressant maintenance therapy in China. The probable cause is overstating not only the severity of side effects of medication but also the mild course of disease progression. Future research on disease course will help determine the length of maintenance treatment time.
Our findings highlight the need of searching for serum AQP4-IgG even in patients with recurrent SPTM and suggest that NMOSD with recurrent SPTM may be mild in the acute attack phase and have a favorable long-term prognosis. As rituximab is useful for NMOSD and appears somewhat effective for MS, it might be a reasonable choice if uncertain, tests are not yet returned, or AQP4-IgG is not available.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.