
Abstract
Teriflunomide is a once-daily oral immunomodulator approved for relapsing-remitting multiple sclerosis (MS). The objective of this post hoc analysis of the phase 3, pooled TEMSO (NCT00134563) and TOWER (NCT00751881) dataset is to evaluate the effect of teriflunomide treatment on annualised relapse rate and disability worsening across patient subgroups defined according to prior disease-modifying therapy exposure. This analysis provides further supportive evidence for a consistent effect of teriflunomide across a broad range of patients with relapsing MS, including patients who have used and discontinued other disease-modifying therapies.
Introduction
In patients with multiple sclerosis (MS), treatment switches are often considered to improve adherence, address a lack of efficacy or mitigate tolerability and/or safety concerns.1,2 The decision to change therapy is complex, requiring assessment of therapeutic benefit–risk and the potential for further disease worsening. 3 Patients who have previously received treatment and require multiple changes in therapy may have more active disease and be at increased risk of relapse or disease worsening compared with treatment-naïve patients. 4
Teriflunomide is a once-daily oral immunomodulator approved for relapsing-remitting multiple sclerosis (RRMS) that has been evaluated in two pivotal phase 3 studies: TEMSO (NCT00134563) and TOWER (NCT00751881).5,6 In the individual studies and pooled dataset, teriflunomide 14 mg significantly reduced annualised relapse rate (ARR) and risk of disability worsening confirmed for 12 weeks compared with placebo.5,6 In addition, safety and tolerability profiles for teriflunomide were similar within the individual studies and pooled analyses.5–7
TEMSO and TOWER included patients who received one or more other disease-modifying therapies (DMTs) in the 2 years prior to study entry (but had not used them within the 3–6 months before randomisation).5,6,8 We evaluated ARR and disability worsening in patients exposed to prior treatment versus patients who had not received a prior DMT in the previous 2 years, described herein as ‘treatment-naïve’.
Methods
TEMSO and TOWER were multicentre, multinational, randomised (1:1:1 to once-daily oral placebo or teriflunomide 7 or 14 mg), double-blind, parallel-group, placebo-controlled phase 3 studies.5,6 Duration of treatment was fixed in TEMSO (108 weeks), 5 and varied in TOWER (48–173 weeks), where the study ended 48 weeks after last patient randomised. 6 Both studies enrolled adults (18–55 years) with relapsing forms of MS and Expanded Disability Status Scale (EDSS) scores ≤5.5. Patients were required to have ≥1 relapse (12 months) or ≥2 relapses (24 months) before study entry.5,6 Both TEMSO and TOWER included patients who had received one or more DMT in the 2 years prior to study entry but had not used them within 3–6 months before randomisation.5,6,8
Post hoc analyses were performed on the pooled, modified intent-to-treat population from both studies (all patients randomised who received ≥1 dose of study medication), in subgroups defined according to DMT exposure in the previous 2 years: ≥2 prior DMTs, 1 prior DMT or no prior DMT. Reasons for discontinuation or switch of prior therapy were not recorded, but may have been due to perceived sub-optimal treatment response, poor adherence, or safety and tolerability issues. Outcomes included ARR and disability worsening confirmed for 12 weeks (defined as an increase from baseline of ≥1.0 EDSS point (or ≥0.5 points for a baseline EDSS score >5.5) for at least 12 weeks). Magnetic resonance imaging (MRI) was not performed in TOWER and thus is not included as an outcome in this analysis.
Statistical analysis
ARR was derived from an analysis of the number of relapses, using a Poisson regression model with the log of time during treatment as an offset variable. For disability worsening, a log-rank test was used to compare teriflunomide with placebo, and a hazard ratio was estimated using a Cox regression model. For all endpoints, analysis models were adjusted for treatment, EDSS strata at baseline (≤3.5 or >3.5), region, study and prior-treatment subgroup as covariates. Consistency of treatment effect across prior-treatment subgroups was evaluated using a treatment-by-subgroup interaction term. Inferential analyses were performed at the two-sided 5% level of significance.
Results
In total, 2251 patients were included in the pooled analysis. Baseline disease characteristics of the pooled TEMSO/TOWER population according to prior DMT were generally well balanced across the three groups; however, disease duration was shorter in the treatment-naïve population (Table 1). Most patients were treatment-naïve, with ~30% having used one or more prior DMT. Across all treatment groups, the most frequently used prior DMT was interferon beta (IFNβ) (Table 1).
Baseline disease characteristics by number of prior treatments and prior treatment by treatment group (mITT population).
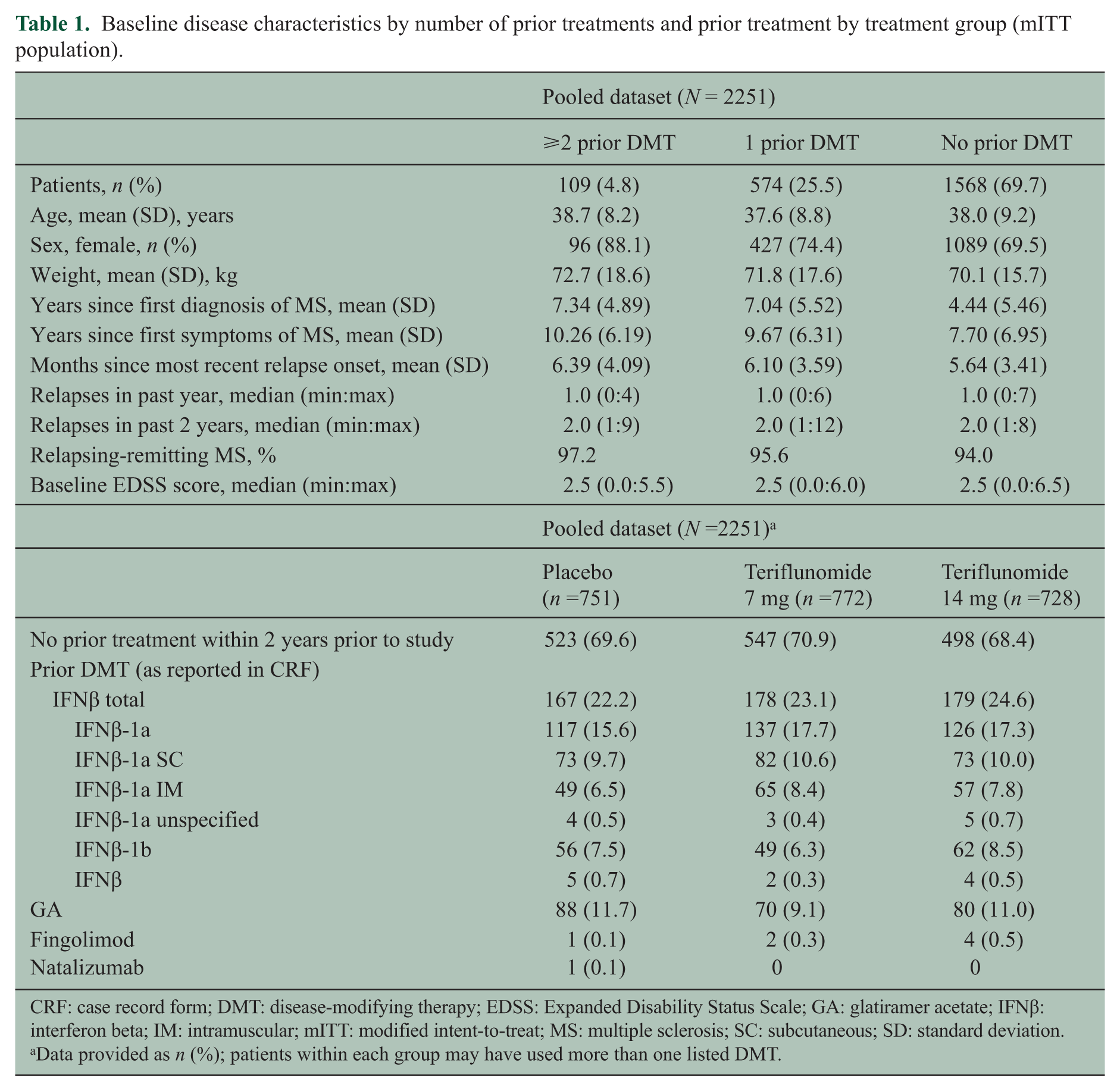
CRF: case record form; DMT: disease-modifying therapy; EDSS: Expanded Disability Status Scale; GA: glatiramer acetate; IFNβ: interferon beta; IM: intramuscular; mITT: modified intent-to-treat; MS: multiple sclerosis; SC: subcutaneous; SD: standard deviation.
Data provided as n (%); patients within each group may have used more than one listed DMT.
ARR
Compared with treatment-naïve patients, ARRs were higher for patients in the placebo group exposed to either ≥2 prior DMTs or 1 prior DMT (Figure 1(a)). Across all subgroups, a greater reduction in ARR was reported following teriflunomide treatment versus placebo. Consistency of treatment effect of teriflunomide 14 mg is supported by the non-significant treatment-by-subgroup interaction (p = 0.4344). Reductions in ARR with teriflunomide were numerically greater in patients who had received ≥2 prior DMTs versus those who had used 1 prior DMT.
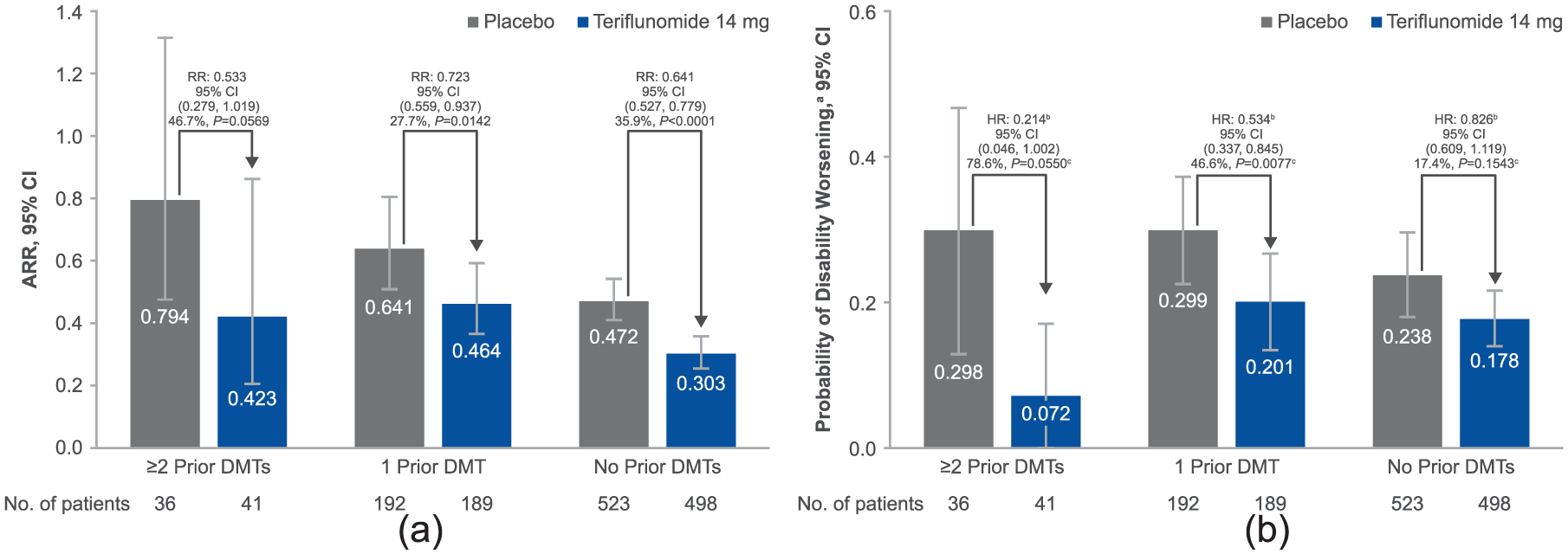
(a) ARR by prior treatment. Overall p value for treatment-by-subgroup interaction for ARR: 14 mg, p = 0.4344. Percentages represent relative risk reductions (95% CI). ARR (95% CI) for teriflunomide 7 mg: ≥2 prior DMTs, 0.463 (0.231, 0.930), RR (95% CI) 0.584 (0.308, 1.104), difference versus placebo 41 .6%, p =0.0977; 1 prior DMT, 0.536 (0.423, 0.680), RR (95% CI) 0.836 (0.643, 1.087), difference versus placebo 16.4%, p =0.1804; no prior DMT, 0.329 (0.285, 0.380), RR (95% CI) 0.698 (0.583, 0.835), difference versus placebo 30.2%, p =0.0001. Overall p value for treatment-by-subgroup interaction for ARR, p =0.3947. In the placebo arms, ARR was significantly higher for patients with ≥2 DMTs (p =0.0183) or 1 prior DMT (p =0.0008) compared with treatment-naïve patients. (b) Disability worsening by prior treatment. aDerived from Kaplan–Meier estimates at week 132. bDerived using a Cox proportional hazard model with treatment, EDSS strata at baseline, region and study as covariates. cDerived from log-rank test, with EDSS strata at baseline, region, study and subgroup as covariates. Overall p value for treatment-by-subgroup interaction for disability worsening: 14 mg, p =0.0697. Percentages represent relative risk reductions (95% CI) on the hazard ratios. Probability of disability worseninga (95% CI) for teriflunomide 7 mg: ≥2 prior DMTs, 0.218 (0.061, 0.376), HR (95% CI) 0.666 (0.223, 1.992), difference versus placebo 33.4%, p =0.5707c; 1 prior DMT, 0.345 (0.259, 0.431), HR (95% CI) 0.950 (0.634, 1.422), difference versus placebo 5.0%, p =0.8505c; no prior DMT, 0.176 (0.139, 0.212), HR (95% CI) 0.792 (0.587, 1.068), difference versus placebo 20.8%, p =0.0948c. Overall p value for treatment-by-subgroup interaction for disability worsening: 7 mg, p =0.6921. In the placebo arms, there was a significantly greater risk of disability progression in patients with 1 prior DMT (p =0.0250) compared with treatment-naïve patients. The risk was also greater in patients with ≥2 prior DMTs, although significance was not reached (p = 0.4486).
Disability worsening
Placebo-treated patients had a numerically greater risk of disability worsening in both prior-treatment subgroups compared with treatment-naïve patients (Figure 1(b)). Consistency of treatment effect of teriflunomide 14 mg was established with a non-significant treatment-by-subgroup interaction, although this trended towards significance (p=0.0697). The treatment effect of teriflunomide 14 mg on reducing risk of disability worsening was numerically greater in patients who had received ≥2 prior DMTs or 1 prior DMT versus treatment-naïve patients.
MRI outcomes (TEMSO only)
As noted for the clinical outcomes, placebo-treated patients in prior DMT groups tended to have higher MRI activity with more enhancing lesions (Supple-mental Figure 1(a)), although teriflunomide positively impacted MRI activity across all subgroups regardless of prior exposure (Supplemental Figure 1(b)).
Discussion
In this analysis of the TEMSO/TOWER pooled data set, teriflunomide 14 mg was associated with reductions in ARR and risk of disability worsening across all subgroups defined by prior DMT exposure compared with placebo. Due to small group sizes limiting statistical power, treatment effect did not reach significance in all instances; however, the direction of change on both outcomes was the same regardless of prior DMT exposure. In the placebo arms, patients with prior DMT exposure were at higher risk of relapses and disability worsening than treatment-naïve patients. Nevertheless, for both ARR and disability worsening, reductions with teriflunomide treatment were numerically greater in patients with prior DMT exposure. These results also help to address prior perceptions that patients who require switching from their current DMT are at higher risk of relapse or disability worsening in the imminent future. 4 This risk was consistently greater in placebo-treated patients who had one or more prior DMT versus treatment-naïve patients.
Although there are limitations to this analysis (e.g. small group sizes and lack of information on reasons for switching from prior DMTs), these pooled subgroup analyses support the efficacy of teriflunomide across a broad range of patients with RRMS, including those who have discontinued previous DMTs – a subgroup of patients that can be challenging to treat.
Footnotes
Acknowledgements
TEMSO and TOWER were conducted in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice and the Declaration of Helsinki. Study protocols were approved by central and local ethics committees and each site’s institutional review board. All patients gave written consent prior to participation. Professor Christian Confavreux (University Claude Bernard Lyon 1, Lyon, France), who passed away in September 2013, was a member of the Steering Committee and contributed to the design and conduct of the TEMSO and TOWER studies, as well as to the analysis and interpretation of the data. This manuscript was reviewed by Larisa Miller and Alex Lublin of Sanofi Genzyme. Editorial assistance was provided by Fiona Woodward of Fishawack Communications Ltd, also funded by Sanofi Genzyme.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship and/or publication of this article: M.S.F. has received research/educational grant support from Bayer HealthCare and Genzyme; honoraria/consulting fees from Bayer HealthCare, Biogen Idec, EMD Canada, Novartis, Sanofi and Teva Canada Innovation; and is a member of company advisory boards/board of directors/or other similar group for Bayer HealthCare, Biogen Idec, Chugai, Merck Serono, Novartis, Opexa Therapeutics, Sanofi and Teva Canada Innovation. J.S.W. has received, within the last year, consulting fees from AbbVie, Alkermes, Bayer HealthCare, Forward Pharma, MedDay, Novartis, Roche Genentech, Sanofi Genzyme, Takeda and Teva. Royalties from the University of Texas Health Science Center at Houston for monoclonal antibodies out-licenced to Chemicon International. G.C. has received compensation in the past year for consulting services and/or speaking activities from Almirall, Biogen, Celgene, Excemed, Forward Pharma, Genzyme, Merck, Novartis, Receptos, Roche, Sanofi and Teva. L.K.’s institution (University Hospital Basel) has received in the last 3 years and used exclusively for research support: steering committee, advisory board and consultancy fees from Actelion, Addex, Bayer HealthCare, Biogen Idec, Biotica, Genzyme, Lilly, Merck, Mitsubishi, Novartis, Ono Pharma, Pfizer, Receptos, Sanofi, Santhera, Siemens, Teva, UCB and XenoPort; speaker fees from Bayer HealthCare, Biogen Idec, Merck, Novartis, Sanofi and Teva; support of educational activities from Bayer HealthCare, Biogen Idec, CSL Behring, Genzyme, Merck, Novartis, Sanofi and Teva; licence fees from Neurostatus Systems GmbH; and grants from Bayer HealthCare, Biogen, Merck, Novartis, Roche, Swiss MS Society, the Swiss National Research Foundation, the European Union and Roche Research Foundations. T.P.O. has received consulting fees and/or research support from Biogen Idec, Merck Serono and Sanofi and has participated in scientific advisory boards and/or speaking activities for Biogen Idec, Merck Serono and Sanofi. A.E.M. has received research support from Biogen Idec, Genentech, Novartis, Questcor, Roche and Sanofi and consulting fees from Accordant Health Services, Acorda Therapeutics, Alkermes, Biogen Idec, EMD Serono, Genentech, Genzyme, GlaxoSmithKline, Mallinckrodt Pharmaceuticals/Questcor, Novartis, Roche and Teva. K.T., M.B. and P.T. are employees of Sanofi Genzyme. P.W.O’C. has received consulting fees and/or research support from Actelion, Bayer, Biogen Idec, BioMS, Cognosci, Daiichi Sankyo, EMD Serono, Genentech, Genmab, Novartis, Roche, Sanofi, Teva and Warburg Pincus.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This study was supported by Sanofi Genzyme.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.