
Abstract
Background:
Open-label 15-month follow-up of the double-blind, placebo-controlled Glatiramer Acetate clinical Trial to assess Equivalence with Copaxone® (GATE) trial.
Objective:
To evaluate efficacy, safety, and tolerability of prolonged generic glatiramer acetate (GTR) treatment and to evaluate efficacy, safety, and tolerability of switching from brand glatiramer acetate (GA) to GTR treatment.
Methods:
A total of 729 patients received GTR 20 mg/mL daily. Safety was assessed at months 12, 15, 18, 21, and 24 and Expanded Disability Status Scale and magnetic resonance imaging (MRI) scans at months 12, 18, and 24. The presence of glatiramer anti-drug antibodies (ADAs) was tested at baseline and months 1, 3, 6, 9, 12, 18, and 24.
Results:
The mean number of gadolinium-enhancing lesions in the GTR/GTR and GA/GTR groups was similar at months 12, 18, and 24. The change in other MRI parameters was also similar in the GTR/GTR and GA/GTR groups. The annualized relapse rate (ARR) did not differ between the GTR/GTR and GA/GTR groups, 0.21 and 0.24, respectively. The incidence, spectrum, and severity of reported adverse events did not differ between the GTR/GTR and GA/GTR groups. Glatiramer ADA titers were similar in the GTR/GTR and GA/GTR groups.
Conclusion:
Efficacy and safety of GTR is maintained over 2 years. Additionally, switching from GA to GTR is safe and well tolerated.
Introduction
Glatiramer acetate (GA) is a mixture of random polypeptides. Its mechanism of action is not fully elucidated but is postulated to involve effects on both adaptive and innate immune mechanisms. 1 Double-blind, placebo-controlled trials demonstrated that 20 mg/mL GA administered by daily subcutaneous injections reduces clinical relapses and magnetic resonance imaging (MRI) lesion activity in relapsing-remitting multiple sclerosis (RRMS) leading to regulatory approval of GA in multiple sclerosis (MS) treatment.2–4 The rate of conversion to clinically definite MS following a first demyelinating event was also reduced with GA, and GA was approved for this indication. 5 Subsequent clinical trials,6–11 a 15-year open-label follow-up study, 12 and post-marketing clinical experience support the efficacy, safety, and tolerability of GA 20 mg daily. In 2015, the patent for glatiramer acetate expired, and the United States Food and Drug Administration recently approved a generic glatiramer acetate (GTR) based on demonstration of physicochemical equivalence and equivalent biological and immunologic effects in murine experimental autoimmune encephalomyelitis, without clinical testing in patients. 13 In contrast, the European Medicines Agency (EMA) considered glatiramer acetate a complex non-biological product and required clinical trial data as an essential part of the market authorization application for generic versions to enable assessment of efficacy, safety, and tolerability.
The 9-month double-blind Glatiramer Acetate clinical Trial to assess Equivalence with Copaxone® (GATE) in RRMS patients demonstrated equivalent efficacy, safety, and tolerability for generic GA (GTR, Synthon BV) and brand GA (Copaxone®, Teva) treatment. 14 Hence, approval for generic GTR was recently obtained in Europe based on a complete and comprehensive package including comparative quality, preclinical and clinical data. 15
The GATE trial was conducted as a Phase III trial which recruited 796 RRMS patients. 14 All patients completing the double-blind part of the GATE trial on assigned treatment were eligible to continue into a 15-month open-label extension on GTR treatment to evaluate the efficacy, safety, and tolerability of prolonged GTR exposure and to assess whether switching from the regulatory-approved brand GA product to GTR can be performed without impacting safety and efficacy.
Methods
Study design
The 9-month GATE core trial (ClinicalTrials.gov NCT01489254) has been reported in detail. 14 Briefly, the GATE trial was a 9-month randomized, double-blind, placebo-controlled, Phase III clinical trial followed by a 15-month open-label extension (Supplemental Figure e-1). Patients provided written informed consent before undergoing any study-related procedures and were re-consented after the second and each subsequent relapse occurring during the study. The study was conducted in accordance with International Conference on Harmonization Guideline for Good Clinical Practice 16 and principles of the Declaration of Helsinki. 17 Central and local ethics committees and Health Authorities approved the study. A Study Steering Committee collaborated with the sponsor (Synthon BV) to design the study protocol and monitor its conduct. An independent Data and Safety Monitoring Board reviewed trial conduct and safety data. Data were gathered by the investigators and analyzed by the sponsor.
Objectives
The objectives of the open-label extension were to evaluate efficacy, safety, and tolerability of prolonged (2-year) GTR treatment and of switching from GA to GTR. In addition, the formation of glatiramer anti-drug antibodies (ADAs) over the duration of the entire trial was evaluated.
Participants
Patients from 118 sites in 17 countries were enrolled into the double-blind part of the trial between December 2011 and March 2013. Patients eligible for the double-blind study were aged 18–55 years, had RRMS fulfilling the McDonald criteria, 20 Expanded Disability Status Scale (EDSS) score 21 of 0–5.5, at least one documented relapse in the previous year, and 1–15 gadolinium-enhancing lesions on T1-weighted brain MRI at screening. 14 All patients completing the double-blind study on assigned treatment were eligible to continue into the 15-month open-label extension. The last patient completed follow-up of the open-label extension in January 2015.
Randomization
In the double-blind core study, eligible patients were randomly assigned in a 4.3:4.3:1 ratio to receive daily subcutaneous GTR (20 mg/mL), GA (20 mg/mL), or matching placebo (PLC) for 9 months. In the open-label extension, all patients received daily GTR (20 mg/mL) for 15 months.
Procedures
At each study site, a treating neurologist supervised medical management. An examining neurologist determined EDSS scores at scheduled and unscheduled visits. During the open-label extension, the following assessments were performed: safety evaluation at months 12, 15, 18, 21, and 24 and EDSS scoring and brain MRI scans at months 12, 18, and 24. Patients completed a diary for 14 consecutive days at open-label treatment initiation and month 12, recording the presence and intensity of five injection site symptoms (pain, itchiness, redness, swelling, or lumps) as previously reported. 14 Serum samples were collected for assessment of glatiramer ADA at baseline and months 1, 3, 6, 9, 12, 18, and 24 (Supplemental Figure e-1). The presence and titer of glatiramer ADA were analyzed by enzyme-linked immunosorbent assay (ELISA) with biotinylated glatiramer acetate captured on a streptavidin-coated solid phase and polyclonal anti-human Ig as detecting agent. Titers of all samples which were confirmed positive were determined. The bioanalytical method performance was validated according to recent relevant regulatory guidances and white papers. 22 An MS relapse was defined as new or recurring neurological symptoms, without fever or infection, lasting at least 24 hours, and accompanied by new objective neurological findings on the examining neurologist’s evaluation. Safety assessments included monitoring of adverse events, local injection site reactions, vital signs, and laboratory tests. Neurological symptoms related to confirmed relapses and local injection site reactions recorded in the tolerability diaries were not additionally reported as adverse events. An immediate post-injection reaction was defined as a reaction associated with at least one or more of the following symptoms: vasodilatation, chest pain, dyspnea, palpitation, or tachycardia. Standardized brain MRI scans were analyzed centrally by the Image Analysis Center in Amsterdam, the Netherlands.
Statistical analysis
All efficacy and safety analyses were performed using the full analysis and safety populations, respectively (all randomized patients who received at least one study drug injection). The open-label endpoints were not formally tested comparing treatment groups but summarized per treatment group with point estimates and 95% confidence intervals (CIs) using an appropriate covariance model including the stratification variables (geographical region and the number of gadolinium-enhancing MRI lesions at screening) as covariates. SAS® version 9.4 was utilized for statistical analyses.
Results
To enable a comprehensive overview of the clinical course over the entire 24-month exposure period and to facilitate the assessment of potential effects of switching from GA to GTR, the double-blind part and open-label extension of this trial are reported as separate periods in the results tables. However, the emphasis of this paper is results obtained in the open-label extension.
Baseline characteristics and follow-up
A total of 796 patients were randomized and 735 patients completed the 9-month double-blind core trial. 14 A total of 729 patients completed the double-blind part of the GATE trial on assigned treatment, these were eligible to continue into the 15-month open-label extension, 728 patients entered the open-label extension, 324 patients in the GTR/GTR group, 323 in the GA/GTR group, and 81 in the PLC/GTR group. Of 728 patients who entered the open-label extension, 670 (92.0%) completed the trial, with 304 (93.8%) patients in the GTR/GTR group, 300 (92.9%) in the GA/GTR group, and 66 (81.5%) in the PLC/GTR group. The most common reasons for discontinuation were withdrawal of consent (4.1%) and adverse event (1.4%; Figure 1).
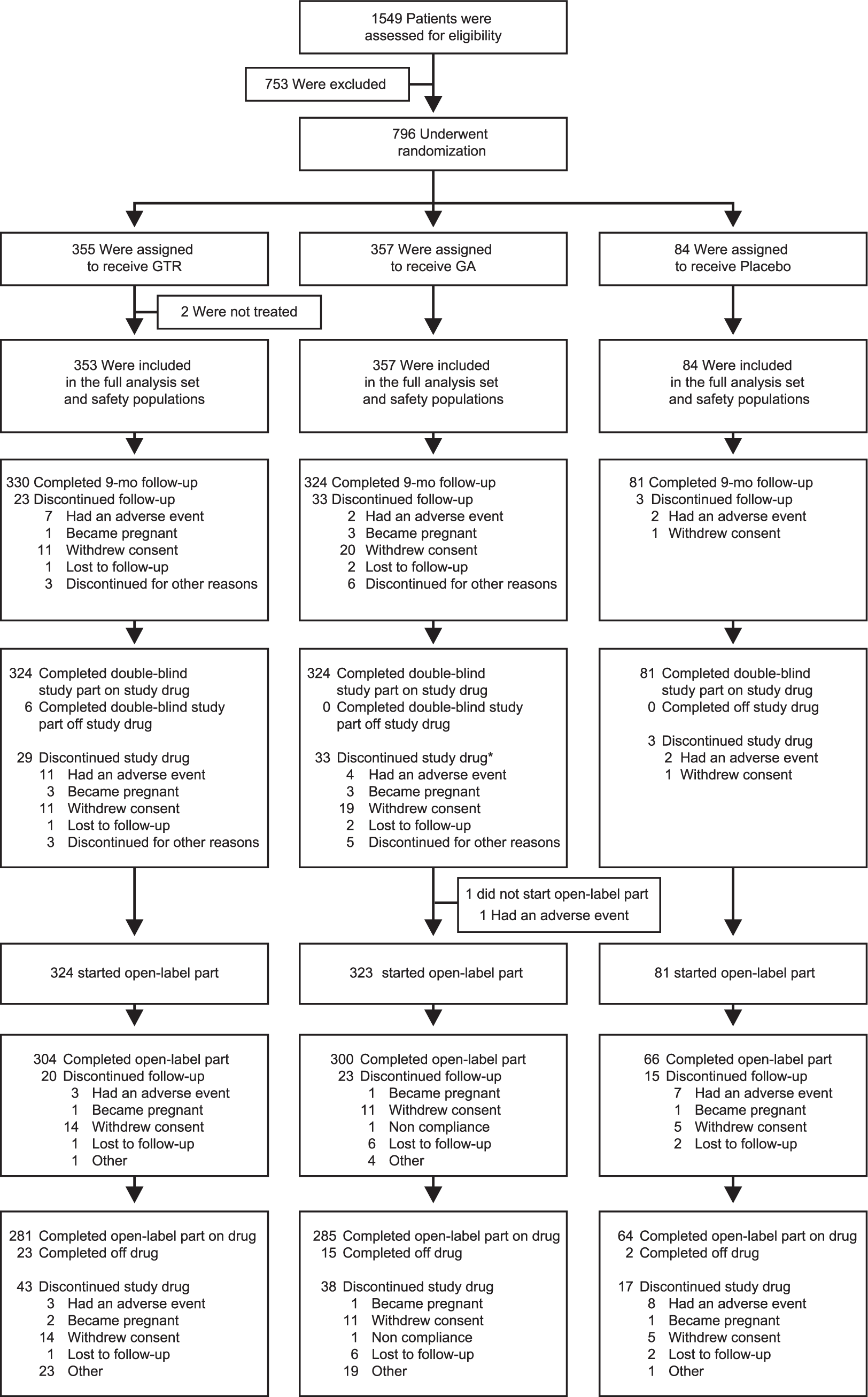
Enrollment and follow-up of study participants.
MRI results
During the open-label extension, the mean numbers of new and persisting gadolinium-enhancing lesions in the GTR/GTR and GA/GTR groups were similar at months 12, 18, and 24 and averaged 0.6–0.7 in both groups. In the PLC/GTR group, the mean number of new and persisting gadolinium-enhancing lesions declined from 1.7 at month 12 to 0.7 at month 18 and 0.9 at month 24 (Figure 2). The change in the number of new T2 lesions reflecting the cumulative lesion load during the extension was similar in the GTR/GTR and GA/GTR groups but higher in the PLC/GTR group (Table 1). Changes in other MRI parameters (T2 lesion volume, T1 hypointense lesion volume, and brain volume) were similar in the GTR/GTR and GA/GTR groups and larger in the PLC/GTR group (Table 1).
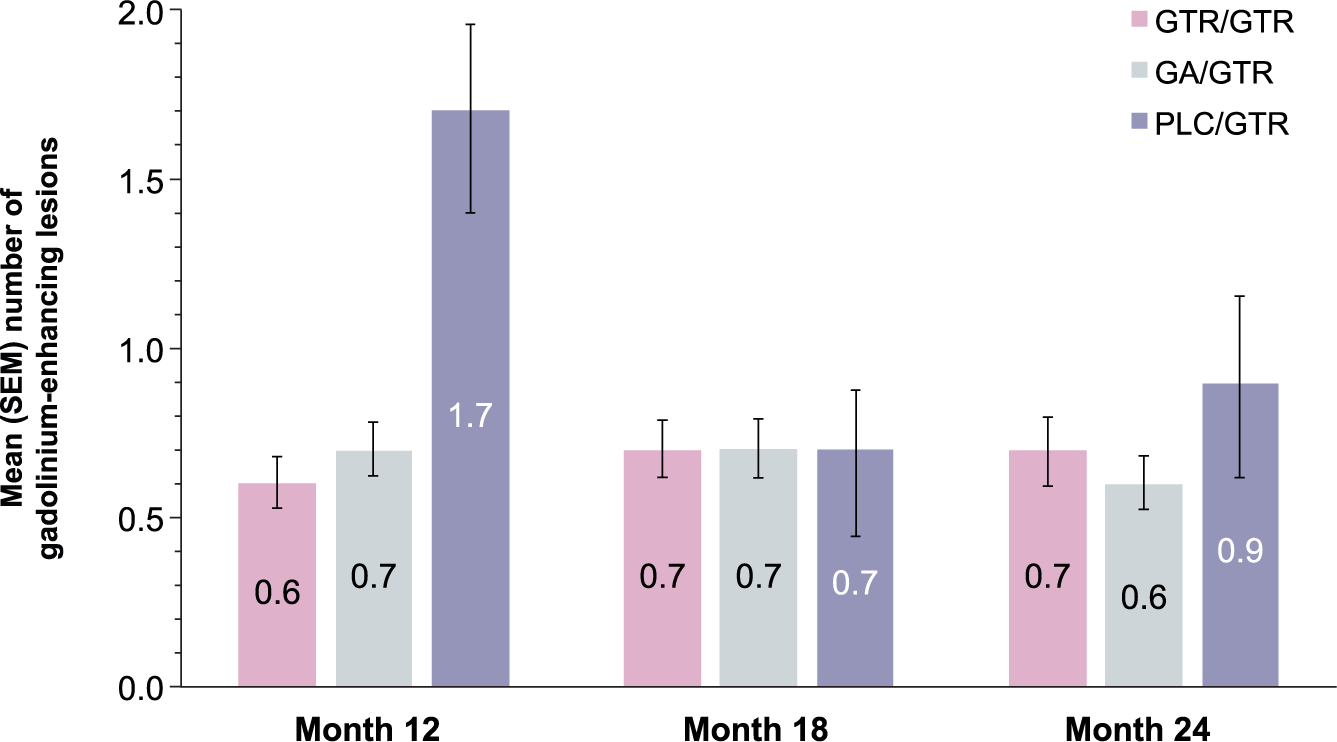
Gadolinium-enhancing lesion number.
MRI and clinical endpoints in double-blind study and open-label extension (full analysis population).
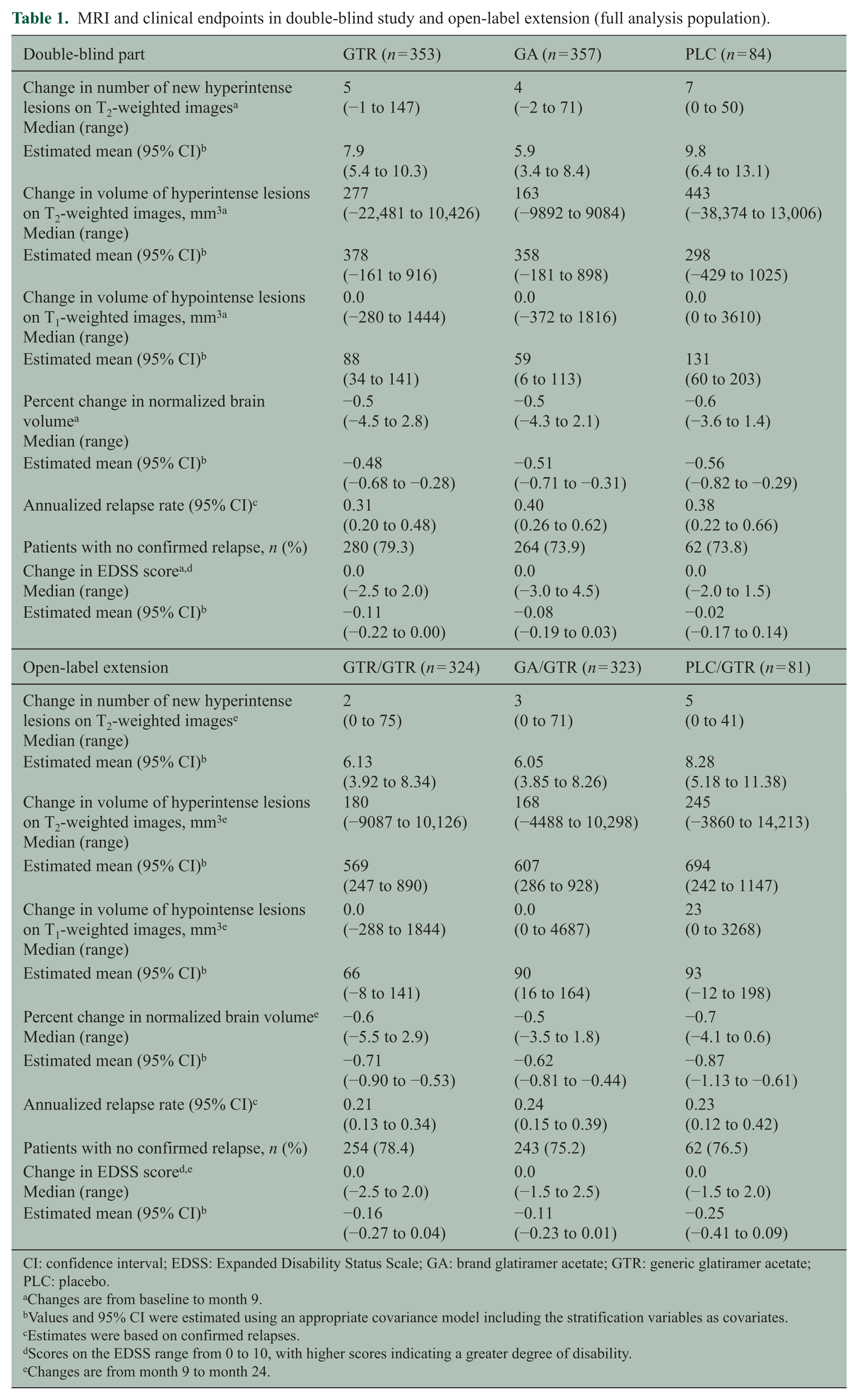
CI: confidence interval; EDSS: Expanded Disability Status Scale; GA: brand glatiramer acetate; GTR: generic glatiramer acetate; PLC: placebo.
Changes are from baseline to month 9.
Values and 95% CI were estimated using an appropriate covariance model including the stratification variables as covariates.
Estimates were based on confirmed relapses.
Scores on the EDSS range from 0 to 10, with higher scores indicating a greater degree of disability.
Changes are from month 9 to month 24.
Clinical outcomes
Estimated annualized relapse rates in the extension study were GTR/GTR: 0.21 (95% CI, 0.13–0.34), GA/GTR: 0.24 (95% CI, 0.15–0.39), and PLC/GTR: 0.23 (95% CI, 0.12–0.42). The percentages of patients free from confirmed relapses were GTR/GTR: 78.4%, GA/GTR: 75.2%, and PLC/GTR: 76.5%. The estimated annualized relapse rate in the GTR/GTR group over the complete 24 months of the trial was 0.25 (95% CI, 0.18–0.37). The median change in EDSS score was 0.0 in all three groups (Table 1).
Tolerability and safety
In the open-label extension, similar proportions of patients reported adverse events in the GTR/GTR (33.3%) and GA/GTR (36.5%) groups compared to 43.2% in the PLC/GTR group (Table 2 and Supplemental Table e-1). Adverse events of severe intensity, serious adverse events, and adverse events leading to discontinuation of study medication and/or trial participation were infrequent and also reported with similar rates in the GTR/GTR and GA/GTR groups. The most common serious adverse events were MS relapse (two patients in the GTR/GTR group and three patients in the GA/GTR group), joint dislocation (one patient in the GTR/GTR and GA/GTR groups), and uterine leiomyoma (two patients in the GA/GTR group). All other serious adverse events occurred in single patients.
Adverse events in double-blind and open-label part (safety population).
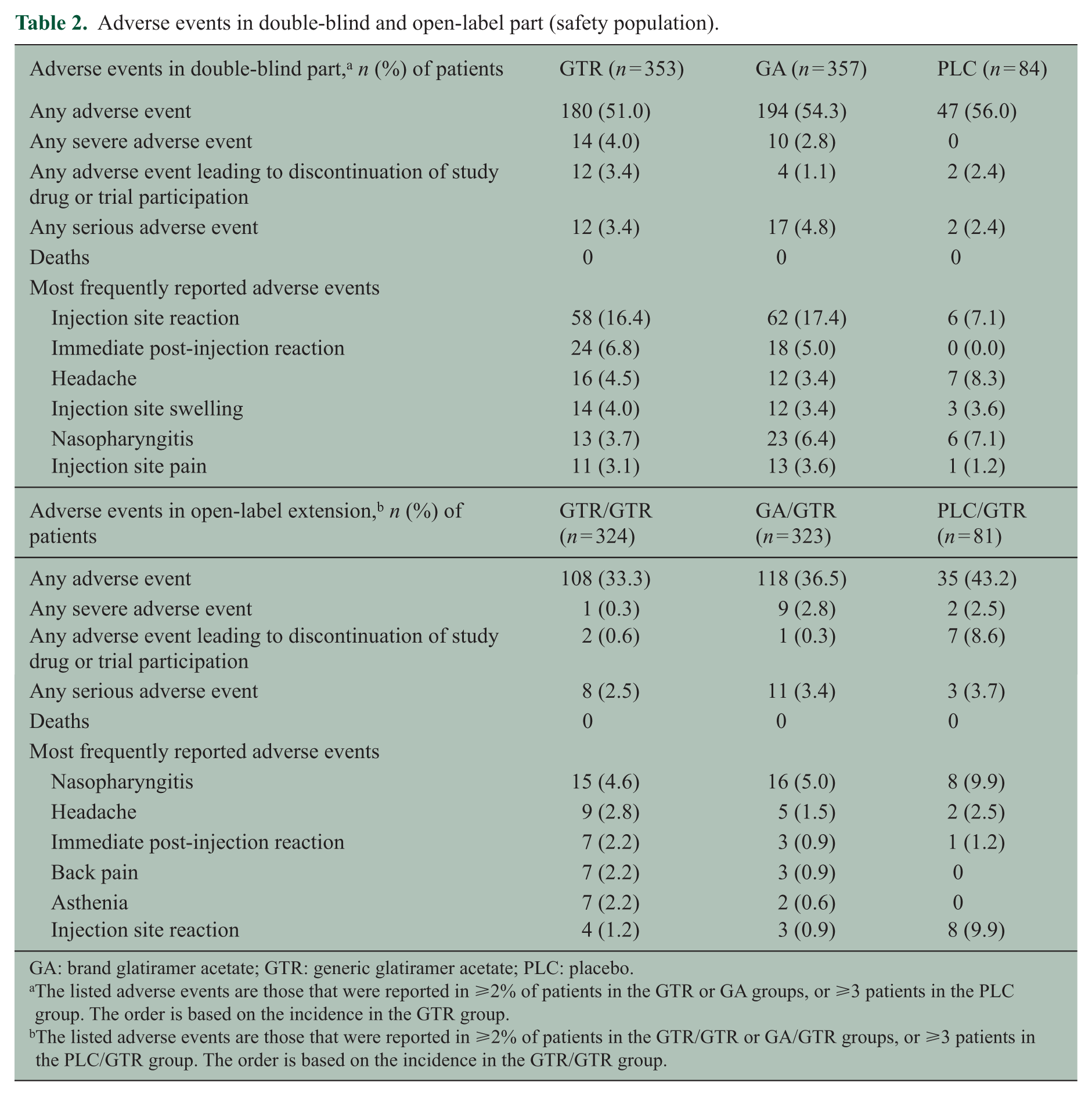
GA: brand glatiramer acetate; GTR: generic glatiramer acetate; PLC: placebo.
The listed adverse events are those that were reported in ⩾2% of patients in the GTR or GA groups, or ⩾3 patients in the PLC group. The order is based on the incidence in the GTR group.
The listed adverse events are those that were reported in ⩾2% of patients in the GTR/GTR or GA/GTR groups, or ⩾3 patients in the PLC/GTR group. The order is based on the incidence in the GTR/GTR group.
Injection site reactions occurred in similar proportions in the GTR/GTR (1.2%) and GA/GTR (0.9%) groups versus 9.9% in the PLC/GTR group. Immediate post-injection reactions occurred in 2.2%, 0.9%, and 1.2% of patients in the GTR/GTR, GA/GTR, and PLC/GTR groups, respectively. Based on the patient’s self-assessment of injection site symptoms, the proportion of patients scoring zero to five symptoms was similar in the GTR/GTR and GA/GTR groups at 5 minutes and 24 hours post injection at months 9 and 12 (Supplemental Figure e-2). Clinically significant vital sign and laboratory abnormalities (blood hematology, biochemistry, and urinalyses) were uncommon in all three treatment groups.
Glatiramer ADAs
Glatiramer ADAs were induced quickly after treatment initiation, and maximum titer levels were detected 3 months after initiation of treatment and gradually declined thereafter but remained detectable up to month 24. During the 9-month double-blind core trial, the formation of glatiramer ADA was comparable with GTR or GA treatment, as reflected by similar number of patients with confirmed positive samples, more than 86% at any given visit in both groups, with comparable serum glatiramer ADA titers. During the open-label extension, the glatiramer ADA titer levels in the group switching from GA to GTR treatment remained comparable to the titer levels in the group continuing on GTR treatment (Figure 3).
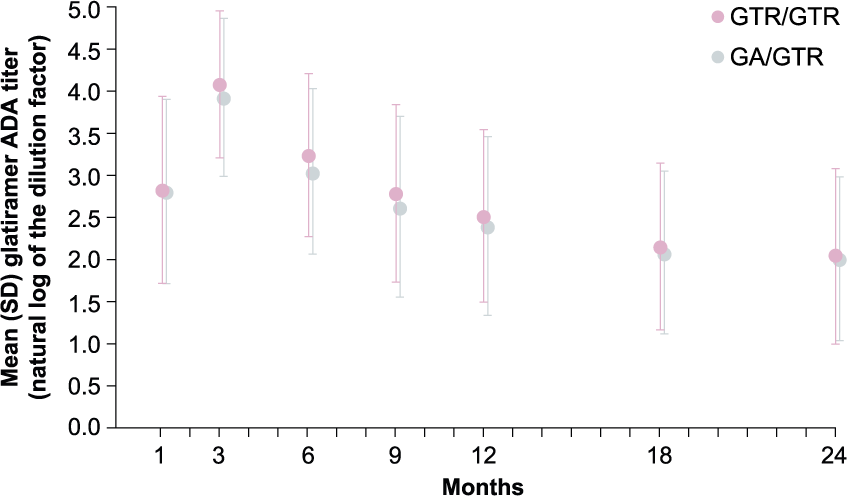
Glatiramer anti-drug antibody titer.
Discussion
Herein, we present results of the 15-month open-label extension of the GATE trial demonstrating that efficacy and safety is maintained over 2 years with GTR. In addition, switching from branded GA to GTR is safe and efficacy is maintained. With the combined 24-month data, the ARR of the group receiving GTR for 24 months was 0.25 which is comparable to the 2-year ARR reported in recent clinical trials with GA.6–8,10,23
Efficacy on MRI was reproduced in patients switching from PLC to GTR including the expected development over time. In the PLC/GTR group, the effect of GTR treatment on the number of new and persisting gadolinium-enhancing and T2 lesions was present from month 18 onwards, that is, 9 months after initiation of GTR treatment, corresponding to previously published data that GA treatment effect on MRI develops over 6 months.3,10 The reduction of MRI activity in GA/GTR and GTR/GTR groups obtained during the double-blind core study continued at similar level during the open-label extension. Thus, switching from branded GA to GTR did not affect efficacy in RRMS patients.
Glatiramer ADAs develop in almost all RRMS patients treated with GA 20 mg administered by daily subcutaneous injection. The levels of glatiramer ADAs peak after 3 months of treatment and remain detectable up to 15 years of continued GA treatment without interfering with its therapeutic efficacy or safety.18,19,24–26 Although glatiramer ADAs have not been shown to impact efficacy or safety of GA treatment, it is expected that a generic version of glatiramer acetate will similarly induce glatiramer ADAs. The GATE trial provided a unique opportunity to directly compare ADA formation patterns as assessed by state-of-the-art methods in the comparative part of the trial and to investigate potential effects on ADA generation after switching from branded GA to generic GTR during the open-label extension. The current trial demonstrates that the incidence and titer of glatiramer ADAs were comparable with GTR and GA treatment. Moreover, switching from GA to GTR did not affect ADA titers. These results demonstrate that branded GA and generic GTR have comparable immunogenicity. To date, this is the only generic version of glatiramer for which immunogenicity has been evaluated in RRMS patients using samples obtained in a comparative trial and analyzed using state-of-the-art ADA assays.
The GATE trial demonstrates the efficacy, safety, and tolerability of GTR in patients with RRMS based on a specifically designed and adequately powered clinical trial. Moreover, the extension part of this trial reported herein demonstrates that patients can be switched safely from branded GA to generic GTR without loss of efficacy, safety, or tolerability. These data should help patients and prescribers to positively consider GTR as an alternative to branded GA.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: K.S. reports grants and personal fees from Synthon during the conduct of the study; grants from Neuron; and grants and personal fees from Roche, Biogen Idec, Novartis, TEVA, Merck, Genzyme, and Receptos outside the submitted work. F.B. reports personal fees from Bayer Schering Pharma, Sanofi Aventis, Biogen Idec, Teva, Merck Serono, Novartis, Roche, Synthon BV, Jansen Research, and Genzyme outside the submitted work. A.N.B. reports no disclosure. C.W. reports personal fees from Synthon BV during the conduct of the study; personal fees from Novartis AG, Teva Ltd, BBB BV, and Desitin GmbH outside the submitted work. E.R.W.v.d.T., J.J.L.O., D.F.E., R.M., and N.P.K. report personal fees from Synthon BV during the conduct of the study and personal fees from Synthon BV outside the submitted work. J.A.C. is a co-editor of Multiple Sclerosis Journal Experimental, Translational and Clinical and reports personal fees from EMD Serono, Genentech, Genzyme, Innate Immunotherapeutics, and Vaccinex outside the submitted work.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.