
Abstract
Background:
Delayed-release dimethyl fumarate (DMF) demonstrated strong efficacy and a favorable benefit–risk profile for patients with relapsing–remitting multiple sclerosis (RRMS) in phase 3 DEFINE/CONFIRM studies. ENDORSE is an ongoing long-term extension of DEFINE/CONFIRM.
Objective:
We report efficacy and safety results of a 5-year interim analysis of ENDORSE (2 years DEFINE/CONFIRM; minimum 3 years ENDORSE).
Methods:
In ENDORSE, patients randomized to DMF 240 mg twice (BID) or thrice daily (TID) in DEFINE/CONFIRM continued this dosage, and those initially randomized to placebo (PBO) or glatiramer acetate (GA) were re-randomized to DMF 240 mg BID or TID.
Results:
For patients continuing DMF BID (BID/BID), annualized relapse rates were 0.202, 0.163, 0.139, 0.143, and 0.138 (years 1–5, respectively) and 63%, 73%, and 88% were free of new or enlarging T2 hyperintense lesions, new T1 hypointense lesions, and gadolinium-enhanced lesions, respectively, at year 5. Adverse events (AEs; serious adverse events (SAEs)) were reported in 91% (22%; BID/BID), 95% (24%; PBO/BID), and 88% (16%; GA/BID) of the patients. One case of progressive multifocal leukoencephalopathy was reported in the setting of severe, prolonged lymphopenia.
Conclusion:
Treatment with DMF was associated with continuously low clinical and magnetic resonance imaging (MRI) disease activity in patients with RRMS. These interim data demonstrate a sustained treatment benefit and an acceptable safety profile with DMF.
Keywords
Introduction
Delayed-release dimethyl fumarate (DMF) is an oral treatment for patients with relapsing–remitting multiple sclerosis (RRMS).1,2 In two 2-year pivotal phase 3 trials (DEFINE and CONFIRM) in patients with RRMS, DMF significantly reduced clinical and magnetic resonance imaging (MRI) activity and demonstrated an acceptable safety profile.3,4 ENDORSE is an ongoing 12-year extension of DEFINE/CONFIRM designed to evaluate the long-term efficacy and safety of DMF. We report a 5-year interim analysis (2 years DEFINE/CONFIRM; 3 years ENDORSE) of clinical and MRI outcomes and safety from ENDORSE. This report focuses on data for DMF 240 mg twice daily (BID; the approved dosage); however, data for all treatment groups are presented in figures or tables.
Methods
Patients and study design
In DEFINE/CONFIRM, eligible patients were of age 18–55 years, had a diagnosis of RRMS, 5 an Expanded Disability Status Scale (EDSS) 6 score of 0–5.0, and ⩾1 relapse within 1 year before randomization or ⩾1 gadolinium-enhanced (Gd+) lesion 0–6 weeks before randomization. Key exclusion criteria included relapse or corticosteroid treatment within 50 days before randomization or prior treatment with glatiramer acetate (GA) within 3 months before randomization (DEFINE) or at any time (CONFIRM). Patients were randomized to DMF 240 mg BID or thrice daily (TID) or placebo (PBO; 1:1:1 in DEFINE) or daily GA 20 mg (1:1:1:1 in CONFIRM) for 96 weeks.3,4
ENDORSE enables up to 14 years of follow-up (2 years DEFINE/CONFIRM + 12-year extension; Figure 1). Originally designed as a multicenter, randomized, dose-blind, dose-comparison study, patients who received DMF 240 mg BID or TID in either parent study remained on the same dosage in ENDORSE. Patients who received PBO or GA were randomized 1:1 to DMF 240 mg BID or TID. After initiation of ENDORSE, DMF was approved for RRMS in several countries at 240 mg BID. A protocol amendment (approved March 2014) outlines a second, open-label phase (beyond year 5), in which all participants receiving DMF 240 mg TID are switched to BID dosing.
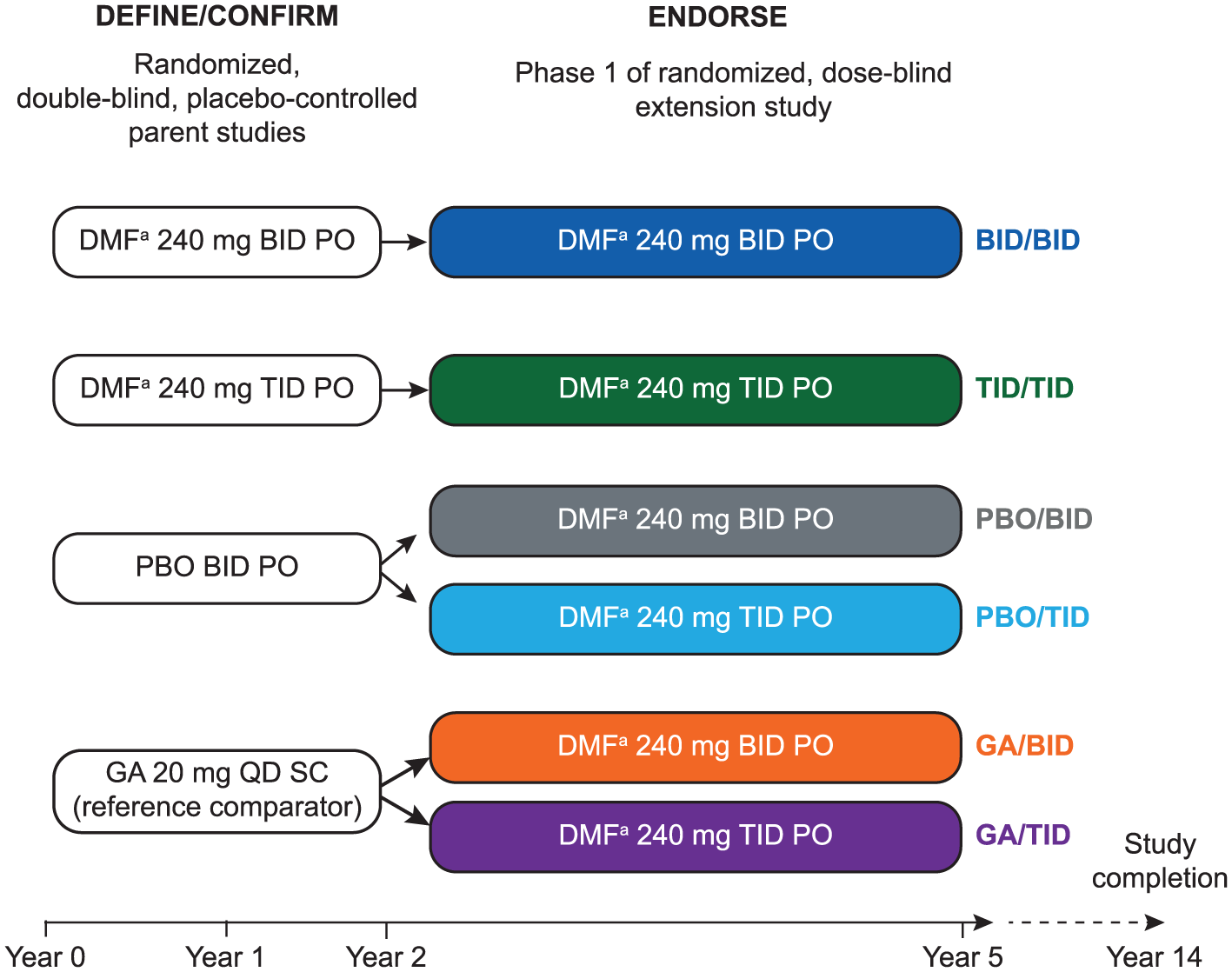
Design of ENDORSE extension study (phase 1).
ENDORSE enrolled eligible patients who completed DEFINE/CONFIRM, excluding those who experienced significant changes in medical history, withdrew consent; discontinued study treatment; or if alanine transaminase (ALT), aspartate aminotransferase (AST), or gamma-glutamyl transpeptidase increased to >3 times the upper limit of normal (ULN). The final (week 96) visit of DEFINE/CONFIRM served as the baseline for ENDORSE; patients were followed every 4 weeks for 24 weeks and every 12 weeks thereafter for up to 12 years.
Efficacy assessments
The primary efficacy endpoint was the proportion of patients relapsed at 2 years (DEFINE) and annualized relapse rate (ARR) at 2 years (CONFIRM). Additional endpoints included time to 12-week sustained disability progression and number of new T1 hypointense lesions (T1), new or enlarging T2 hyperintense lesions (T2), and Gd+ lesions at 2 years. Relapse (confirmed by an Independent Neurologic Evaluation Committee) was defined as new or recurrent neurologic symptoms lasting ⩾24 hours, accompanied by new objective neurologic findings.
Secondary objectives of ENDORSE include assessment of long-term ARR, proportion of patients relapsed, disability progression (measured every 6 months by EDSS), and MRI assessments of brain lesions. Patients at sites with validated MRI capability were eligible to participate in the MRI portion of DEFINE/CONFIRM and could continue in the MRI cohort at the same ENDORSE site.3,4 MRI scans were performed yearly for each patient by the same reading center as that of the parent study. MRI endpoints included number of T1, T2, and Gd+ lesions and percentage of patients free of these lesions. Normalized brain volume was determined at baseline of DEFINE/CONFIRM and ENDORSE, and percent brain volume change (PBVC) was calculated automatically for each post-baseline MRI visit relative to baseline.
Safety assessments
The primary objective of ENDORSE was evaluation of long-term safety of DMF in patients with RRMS. Adverse events (AEs) and concomitant medications were monitored and recorded continuously. Laboratory assessments were performed on a schedule: blood chemistry and urinalysis at baseline, every 4 weeks until week 24, and every 12 weeks thereafter and hematological parameters at baseline and every 12 weeks for up to 12 years. On initiation of the amended protocol, the frequency of some study procedures was decreased to every 24 weeks; however, patients continued visits every 12 weeks for drug dispensing and vital signs assessment.
Patients who completed or discontinued DMF and had a lymphocyte count less than the lower limit of normal (LLN) were followed at least every 12 weeks until lymphocyte counts recovered or until 48 weeks after the last dose (whichever came sooner). Unscheduled relapse assessment was performed as necessary.
Statistical analysis
This 5-year interim analysis (data cutoff date: 14 May 2014) included patients who received ⩾1 dose of DMF in ENDORSE. Results are summarized throughout DEFINE/CONFIRM (years 1–2) and ENDORSE (years 3–5). Data are presented according to treatment received in the parent or extension study: continuing DMF (BID/BID and TID/TID) and new to DMF (PBO/BID, PBO/TID, GA/BID, and GA/TID). To increase sample size in the brain atrophy analysis, DMF BID/TID dosing was pooled from the groups new to DMF.
A Poisson or negative binomial regression model was used to analyze ARR. The proportion of patients relapsed or with progression was estimated based on the Kaplan–Meier product limit method. Disability progression was defined as ⩾1.0-point increase in EDSS from baseline EDSS = 1.0 sustained for 24 weeks or ⩾1.5-point increase in EDSS from baseline EDSS = 0 sustained for 24 weeks. Numbers of T1 and T2 lesions were analyzed by negative binomial regression model, adjusted for region and lesion volume at DEFINE/CONFIRM baseline. Number of Gd+ lesions was analyzed by logit regression.
Comparisons of brain atrophy between BID/BID and PBO/DMF and GA/DMF were based on the analysis of covariance of ranked data, adjusted for DEFINE/CONFIRM or ENDORSE baseline number of Gd+ lesions and T2 lesion volume.
No sample size was calculated for ENDORSE; number of eligible patients was determined by the number of DEFINE/CONFIRM participants.
Safety parameters were tabulated according to the treatment received during parent study or extension phase 1, continuing DMF (BID/BID and TID/TID) and new to DMF (PBO/BID, PBO/TID, GA/BID, and GA/TID), and summarized using descriptive statistics.
Standard protocol approvals, registrations, and patient consents
The study was approved by central and local ethics committees and conducted in accordance with International Conference on Harmonization Guidelines for Good Clinical Practice and the Declaration of Helsinki. All patients provided written informed consent.
Results
Efficacy data are described below for the DMF BID dosage and reported for DMF TID in tables or figures; safety data for both dosages are summarized below.
Patients
Of 2651 patients randomized and dosed in DEFINE/CONFIRM, 2079 completed these studies and 1736 were enrolled and dosed in ENDORSE (intention-to-treat (ITT) population): BID/BID, n = 501; TID/TID, n = 502; PBO/BID, n = 249; PBO/TID, n = 248; GA/BID, n = 118; and GA/TID, n = 118. As of 14 May 2014, total follow-up for this 5-year interim analysis was 4981 patient-years. Follow-up of patients continuing and new to DMF was 3058 and 1923 patient-years, respectively. For BID/BID patients remaining on study (n = 364), minimum follow-up was ~5 years. Among patients new to DMF BID in ENDORSE, minimum follow-up for those remaining on study (n = 163) was ~3 years (Supplementary Table e-1). Of the DEFINE/CONFIRM MRI cohort (n = 1221), 746 were treated in ENDORSE: 363 received DMF BID and 383 DMF TID. Patient disposition is presented in Figure 2. Baseline demographic and disease characteristics at the start of DEFINE/CONFIRM were generally well balanced across treatment groups and were similar between the ENDORSE ITT population (Table 1) and MRI cohort (Supplementary Table e-2).
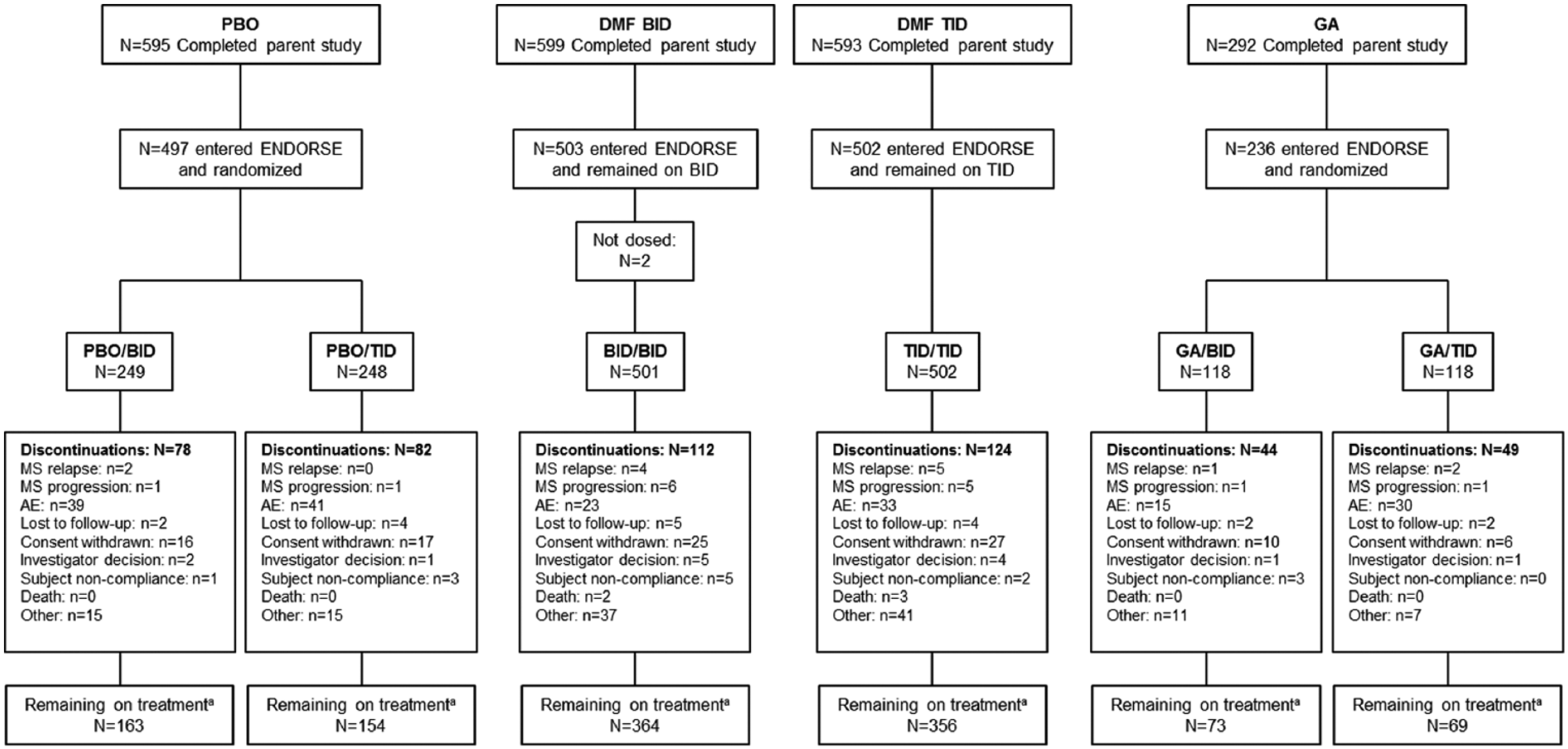
Patient disposition.
Baseline demographics of the ITT population in ENDORSE and at start of DEFINE and CONFIRM.
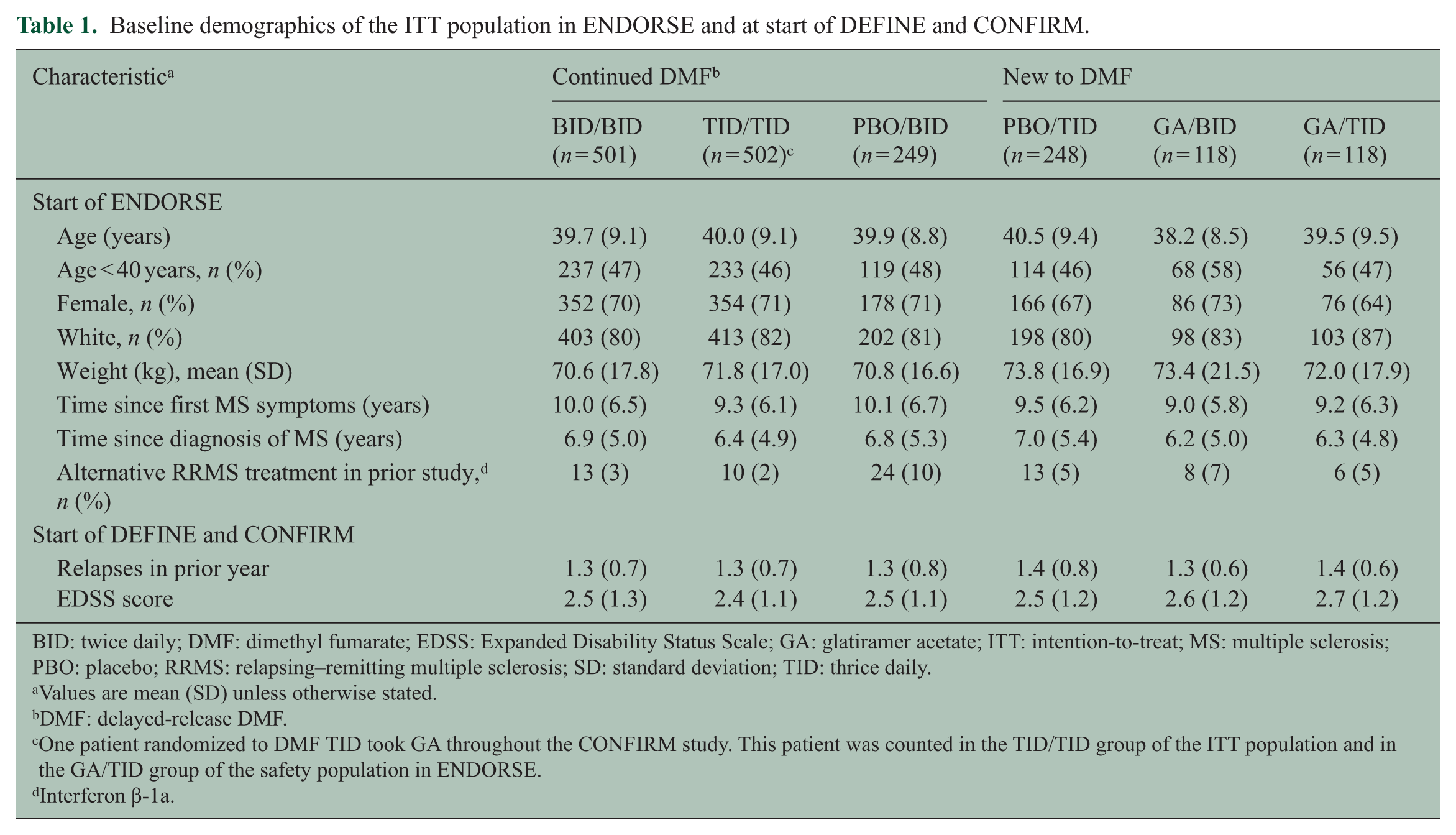
BID: twice daily; DMF: dimethyl fumarate; EDSS: Expanded Disability Status Scale; GA: glatiramer acetate; ITT: intention-to-treat; MS: multiple sclerosis; PBO: placebo; RRMS: relapsing–remitting multiple sclerosis; SD: standard deviation; TID: thrice daily.
Values are mean (SD) unless otherwise stated.
DMF: delayed-release DMF.
One patient randomized to DMF TID took GA throughout the CONFIRM study. This patient was counted in the TID/TID group of the ITT population and in the GA/TID group of the safety population in ENDORSE.
Interferon β-1a.
Efficacy
Relapses
Cumulative ARR for ENDORSE BID/BID patients during years 0–5 was 0.163 (95% confidence interval (95% CI): 0.140, 0.190; Figure 3(a) presents ARRs by yearly interval), and the estimated proportion relapsed at 5 years was 40.1% (95% CI: 35.9%, 44.7%; Figure 3(b)).
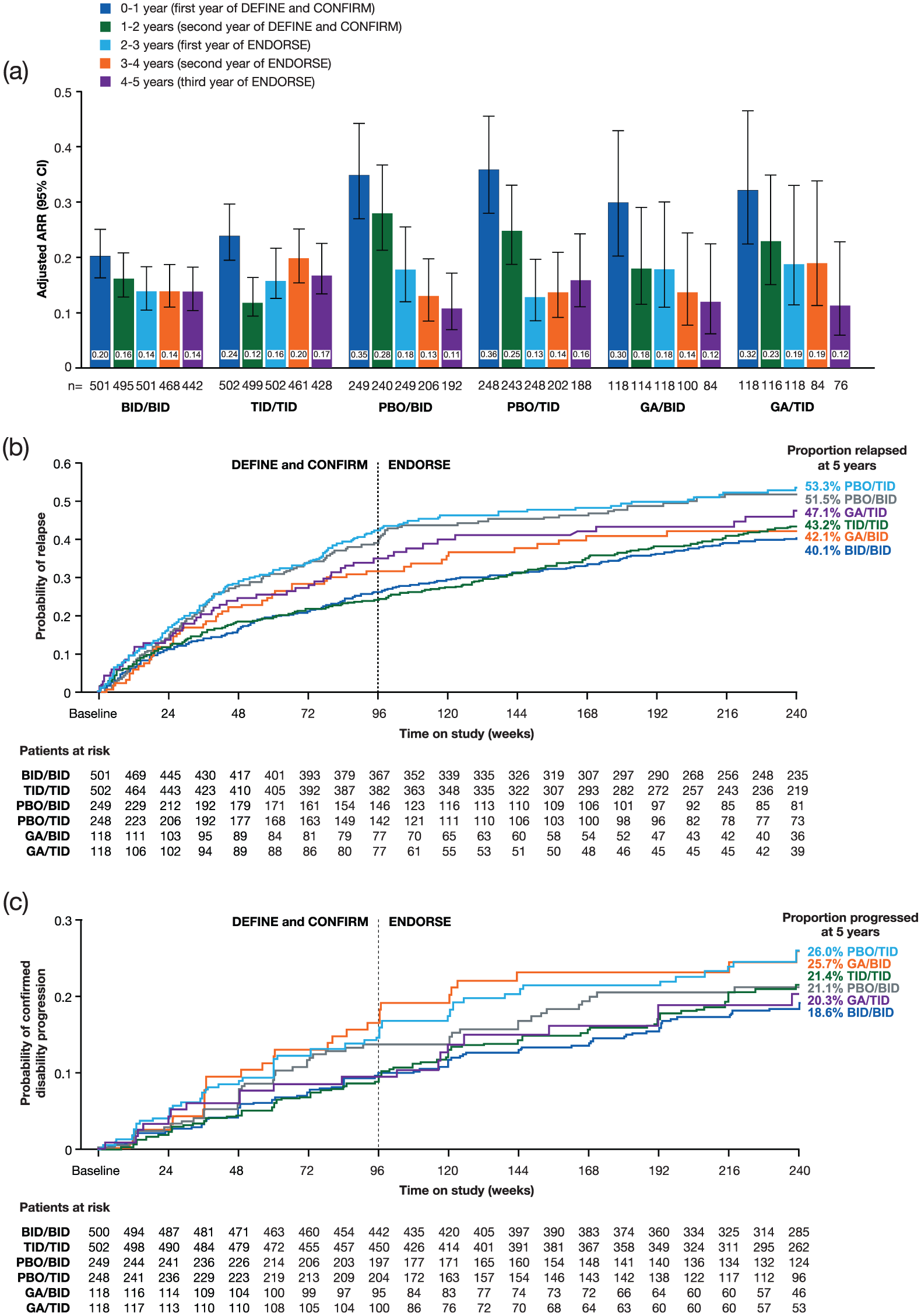
(a) ARR by yearly interval, (b) time to first relapse, and (c) time to disability progression by EDSS (24-week confirmation): DEFINE, CONFIRM, and ENDORSE integrated analysis (ENDORSE ITT population).
Cumulative ARR for ENDORSE PBO/BID patients during years 0–5 was 0.240 (95% CI: 0.196, 0.296). Improvements were generally observed following the switch from PBO to DMF after year 2 (Figure 3(a)). The estimated proportion of PBO/BID patients relapsed at 5 years was 51.5% (95% CI: 45.2%, 58.1%; Figure 3(b)).
Cumulative ARR for ENDORSE GA/BID patients during years 0–5 was 0.199 (95% CI: 0.148, 0.269; Figure 3(a) presents data by yearly interval), and the estimated proportion relapsed at 5 years was 42.1% (95% CI: 33.5%, 52.0%; Figure 3(b)).
Disability progression
An estimated 18.6% (95% CI: 15.3%, 22.4%) of ENDORSE BID/BID patients had confirmed 24-week EDSS progression after 5 years (Figure 3(c)). For PBO/BID patients, the estimated proportion with disability progression after 5 years was 21.1% (95% CI: 16.2%, 27.1%; Figure 3(c)); for GA/BID patients, the corresponding proportion was 25.7% (95% CI: 18.4%, 35.2%; Figure 3(c)).
MRI outcomes
Patients continuing DMF in ENDORSE
Among ENDORSE BID/BID patients, 73% and 63% were free of T1 and T2 lesions, respectively, during years 4–5; 88% were free of Gd+ lesions (year 5 scan). For BID/BID patients, adjusted mean number of T1 and T2 lesions during years 4–5 was 0.5 (95% CI: 0.3, 0.7) and 1.2 (95% CI: 0.8, 1.8), respectively (Figure 4(a) and (b)); mean (±standard error (SE)) number of Gd+ lesions at year 5 was 0.2 ± 0.05 (Figure 4(c)).
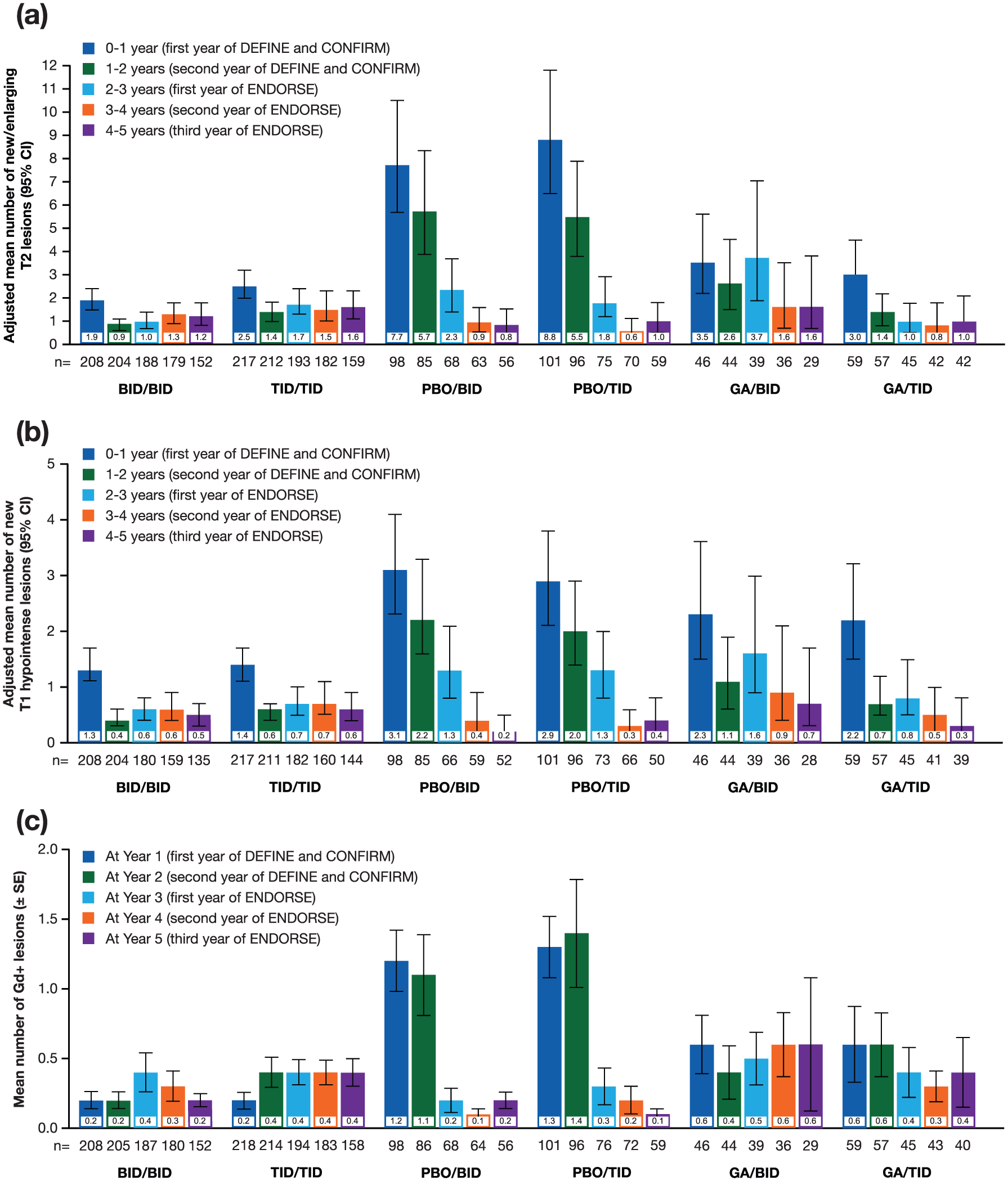
(a) Number of new or enlarging T2 hyperintense lesions by yearly interval, (b) number of new nonenhancing T1 hypointense lesions by yearly interval, and (c) mean number of Gd+ lesions by yearly interval: DEFINE, CONFIRM, and ENDORSE analysis (MRI cohort).
Patients new to DMF in ENDORSE
Of ENDORSE PBO/BID patients, 85% and 68% were free of T1 and T2 lesions, respectively, during years 4–5; 82% were free of Gd+ lesions (year 5 scan). For PBO/BID patients, adjusted mean number of T1 and T2 hyperintense lesions during years 4–5 was 0.2 (95% CI: 0.1, 0.5) and 0.8 (95% CI: 0.4, 1.5), respectively (Figure 4(a) and (b)); mean (±SE) number of Gd+ lesions at year 5 was 0.2 ± 0.06 (Figure 4(c)).
Of ENDORSE GA/BID patients, 64% and 62% were free of T1 and T2 lesions, respectively, during years 4–5 and 86% were free of Gd+ lesions (year 5 scan). For GA/BID patients, adjusted mean number of T1 and T2 lesions during years 4–5 was 0.7 (95% CI: 0.3, 1.7) and 1.6 (95% CI: 0.7, 3.8), respectively, and mean (±SE) number of Gd+ lesions at year 5 was 0.6 ± 0.48.
Brain atrophy
At year 2 of DEFINE/CONFIRM, among patients in ENDORSE, adjusted PBVC from baseline was significantly lower with DMF BID versus PBO (p = 0.0070); in post hoc exploratory analyses, significantly lower PBVC was observed versus GA (p = 0.0035; Table 2). Adjusted PBVC relative to ENDORSE baseline at years 3, 4, and 5 was not significantly different in BID/BID patients compared with the PBO/DMF or GA/DMF groups (Table 2). Annualized rate of adjusted mean PBVC calculated throughout 5 years of follow-up was −0.32 per year (95% CI: −0.37, −0.27)) in BID/BID patients, comparable with that of healthy volunteers. 7
Percent brain volume change.
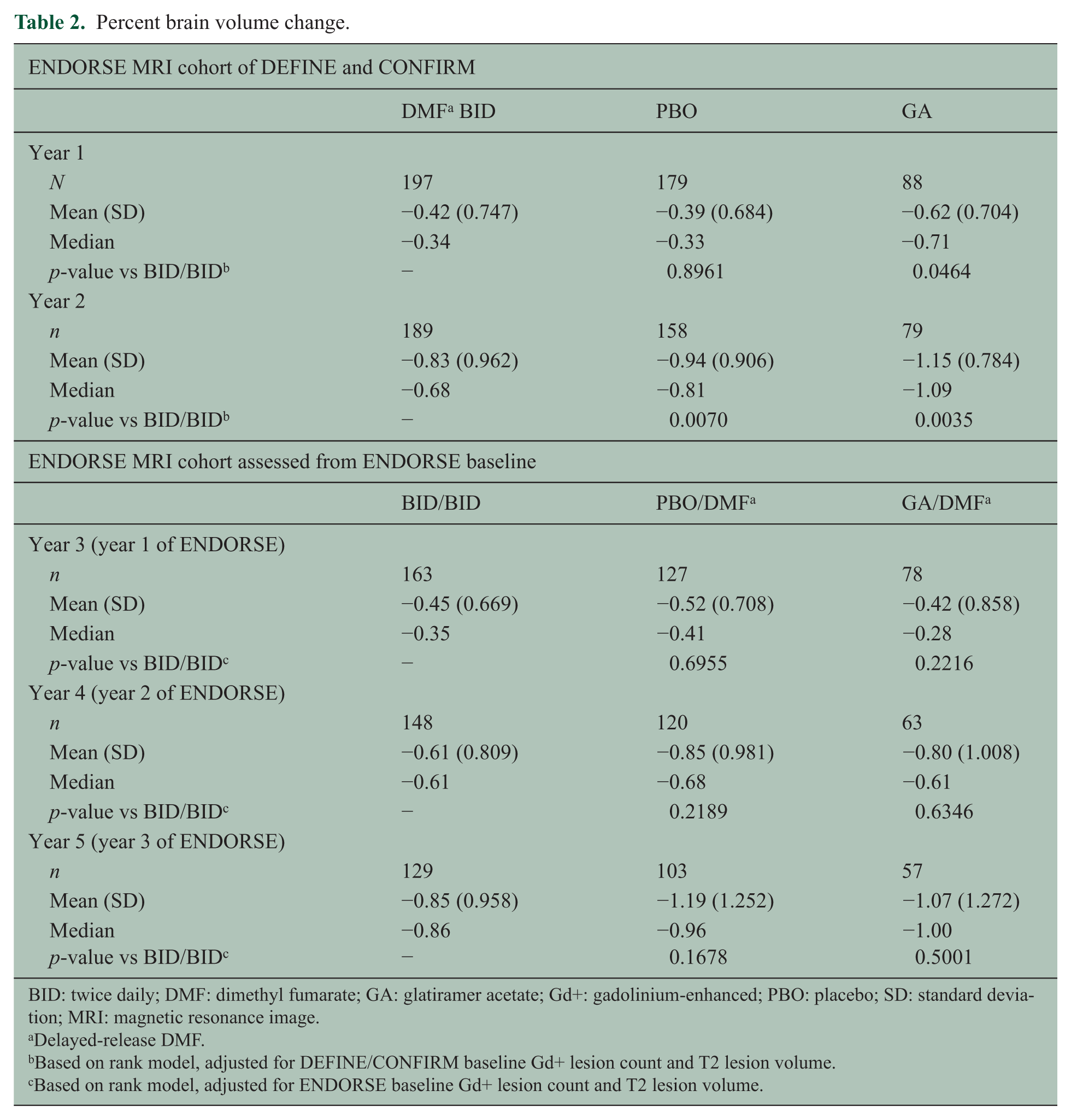
BID: twice daily; DMF: dimethyl fumarate; GA: glatiramer acetate; Gd+: gadolinium-enhanced; PBO: placebo; SD: standard deviation; MRI: magnetic resonance image.
Delayed-release DMF.
Based on rank model, adjusted for DEFINE/CONFIRM baseline Gd+ lesion count and T2 lesion volume.
Based on rank model, adjusted for ENDORSE baseline Gd+ lesion count and T2 lesion volume.
Safety
The overall incidence of AEs, serious AEs (SAEs), and discontinuations due to AEs (Supplementary Table e-3) was similar among the treatment groups who continued DMF from DEFINE/CONFIRM and those new to DMF; however, a higher proportion of patients new to DMF discontinued due to AEs, largely from flushing and gastrointestinal (GI) events that tend to occur early in DMF therapy.3,4,8 The most common individual AEs and SAEs are summarized in Table 3. Multiple sclerosis (MS) relapse and nasopharyngitis were most common in patients continuing DMF. Flushing and GI-related events were more common among patients new to DMF, with incidences highest during the first year of ENDORSE (Supplementary Figure e-1) and generally consistent with those of DMF-treated patients in the parent studies, wherein incidences were highest during the first month and decreased substantially thereafter.3,4,8 Although the study was not designed to assess the duration of each GI or flushing episode in a precise manner, AE duration was summarized based on the investigator-reported onset and resolution dates. There were no apparent differences in the duration of flushing or GI-related AEs across the treatment groups based on the available investigator-reported data. Overall, the median (25%, 75% percentile) duration of flushing and related symptoms (n = 689 events out of 466 patients who had any events) was 80 days (7, 716 days), and the median duration of GI-related events (n = 1011 events out of 523 patients who had any events) was 12 days (4, 71 days).
Most common AEs (incidence ⩾10% in any treatment group) and serious AEs (incidence ⩾3 patients in any treatment group).
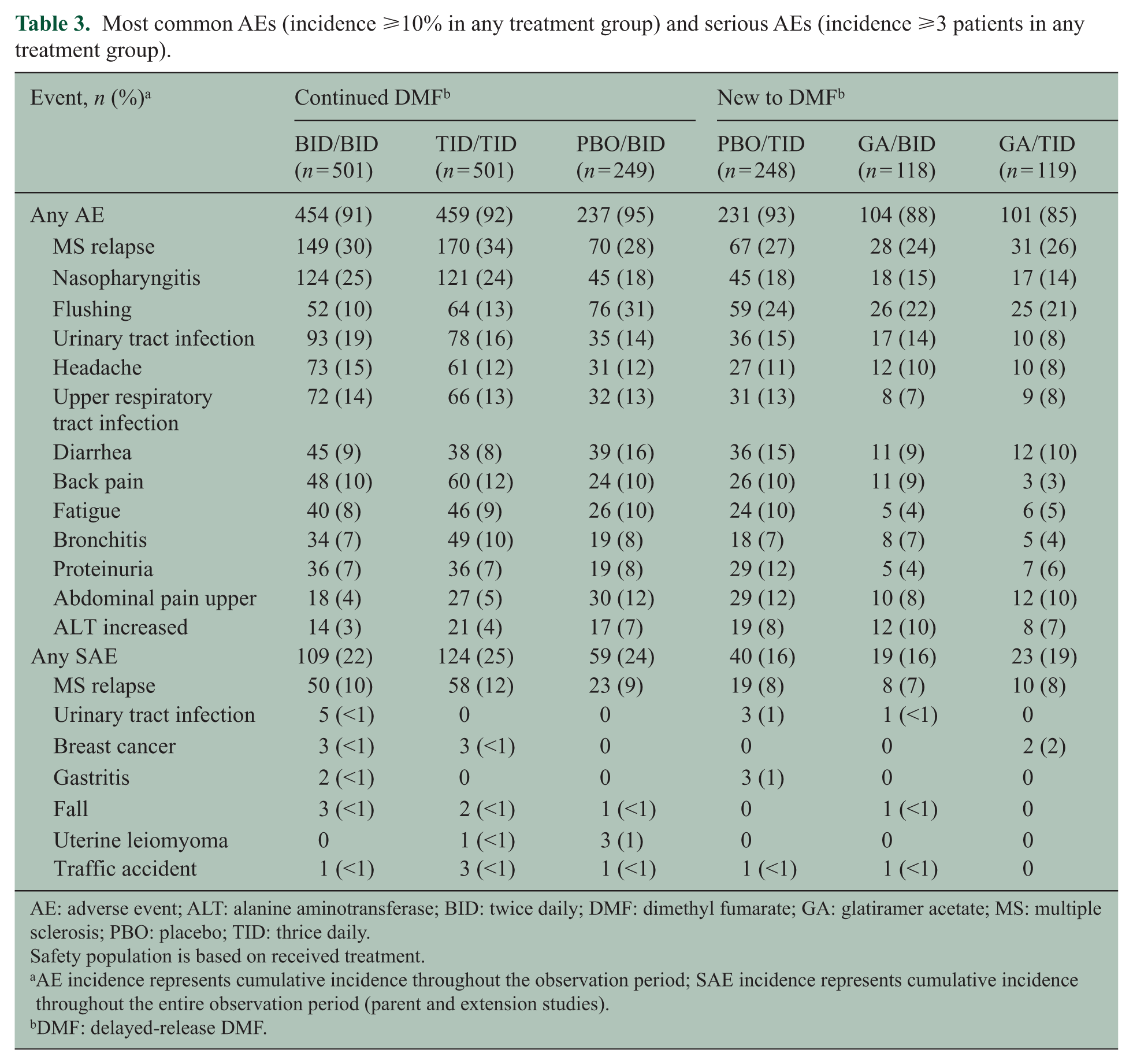
AE: adverse event; ALT: alanine aminotransferase; BID: twice daily; DMF: dimethyl fumarate; GA: glatiramer acetate; MS: multiple sclerosis; PBO: placebo; TID: thrice daily.
Safety population is based on received treatment.
AE incidence represents cumulative incidence throughout the observation period; SAE incidence represents cumulative incidence throughout the entire observation period (parent and extension studies).
DMF: delayed-release DMF.
The incidence of individual AEs leading to treatment discontinuation was ⩽1%–4% (Supplementary Table e-4). Of patients new to DMF, AEs leading to treatment discontinuation were generally related to flushing and GI tolerability; the majority of discontinuations occurred during the first 6 months of treatment (Supplementary Table e-3), consistent with observations in DEFINE/CONFIRM.3,4
The most commonly reported ENDORSE SAE was MS relapse, with other SAEs occurring in ⩽5 patients in any treatment group (Table 3). Incidence of serious infections was ⩽4% in any treatment group (Supplementary Table e-3). A total of 27 malignancies occurred in 18 patients continuing DMF and 8 new to DMF; overall incidence of malignancies was 2% for patients who continued DMF (Supplementary Table e-5). No increased risk of malignancy was observed among DMF-treated patients compared with cancer rates reported for the general MS population (4% summary estimate (95% CI: 3%, 6%)). 9
Hematological findings in ENDORSE were consistent with those from DEFINE/CONFIRM. Patients new to DMF had decreases in mean white blood cell (WBC) and lymphocyte counts, whereas these values remained stable with no further overall decrease in patients continuing DMF. The incidence of lymphocyte counts <0.5 × 109/L was 7%–8% of the patients continuing DMF and 6%–9% of those new to DMF. There was no overall increased risk of serious opportunistic infections; however, subsequent to the data cutoff for this report, a fatal case of progressive multifocal leukoencephalopathy (PML) in a patient treated with DMF 240 mg TID was reported in the setting of severe, prolonged lymphopenia (~290–580 cells/mL3 over 3.5 years); full details of this case are reported separately. 10
Hepatic AEs or SAEs occurred in ⩽3% or <1% of the patients, respectively, in any treatment group. Few patients (⩽3%) continuing DMF had ALT or AST levels ⩾3 × ULN, and no case fulfilled Hy’s law criteria for drug-induced liver injury. The incidence of transaminase elevations in ENDORSE is consistent with observations in DEFINE/CONFIRM, wherein incidences were similar between the DMF- and PBO-treated groups.3,4
In DEFINE/CONFIRM, 19% of the patients receiving DMF BID and 18% of the PBO-treated patients experienced renal or urinary AEs. Renal or urinary events were reported in 23%, 20%, and 13% of ENDORSE BID/BID, PBO/BID, and GA/BID patients, respectively. For BID/BID patients, the most common renal or urinary AEs (⩾3% occurrence in all treatment groups) were proteinuria (7%), microalbuminuria (6%), and hematuria (5%). Renal or urinary AEs resulted in discontinuation in ⩽1% of any treatment group, and serious renal or urinary AEs occurred in <1% of any treatment group.
Discussion
DMF is an oral agent indicated for the treatment of relapsing forms of MS. 11 The availability of agents with novel mechanisms of action affords practitioners a wider array of options to treat RRMS and increases opportunities to individualize therapy for patients intolerant of, or suboptimally responsive to, other therapies. This is particularly germane to MS, the heterogeneous nature of which contributes to variability in therapeutic response.12,13 Additionally, oral agents afford convenience of administration. Efficacy must always be carefully weighed against risk of AEs, particularly throughout longer treatment periods.14–17
In this first phase of ENDORSE, low clinical and MRI activity was sustained during 5 years of DMF treatment. Patients initially randomized to PBO or GA in DEFINE/CONFIRM demonstrated improvements after switching to DMF in ENDORSE; however, with no control group, the relative impact of changes in the natural course of MS in ENDORSE cannot be assessed. In addition, as with other long-term extension studies,17,18 bias could result because not all patients randomized in the parent study were enrolled and dosed in ENDORSE.
The recommended dosage for DMF is 240 mg BID. Data for 240 mg TID were included to explore the general consistency of between-dose effects. In this interim analysis, effect sizes with DMF BID and TID were broadly similar; however, ENDORSE was not powered to analyze dose-dependent differences.
MRI is valuable for diagnosis, prognosis, and assessment of treatment response in patients with MS. 19 Continued DMF treatment resulted in a low frequency of new non-enhancing T1 hypointense lesions, new or enlarging T2 hyperintense lesions, and Gd+ lesions over 5 years. Reduced MRI disease activity was also noted in patients who switched to DMF in ENDORSE.
There was a short delay in treatment initiation in ENDORSE introduced during re-randomization; therefore, some patients who switched from PBO or GA received DMF later than continuously treated patients in the extension (mean treatment gap of 19 days for continuous BID vs 42–65 days for switchers (MRI cohort)).
Accelerated loss of brain tissue in MS correlates with cognitive impairment and worse EDSS and quality of life.20–23 Annual rates of brain volume loss in patients treated continuously with DMF were low and comparable with rates in healthy volunteers. 7 This analysis suggests a continuous beneficial effect of DMF on brain atrophy and a higher impact of early versus delayed treatment. DMF demonstrated improved beneficial effects versus GA; however, statistical significance must be interpreted cautiously given the post hoc, exploratory nature of the comparison. Other disease-modifying MS therapies appear to slow brain atrophy rates;15,24,25 however, comparisons are difficult due to differing study designs, imaging techniques, and analysis methodology across clinical trials.
DMF treatment was associated with relatively low relapse rates, disability progression, and frequency of new MRI lesions over 5 years. Cumulative outcomes generally favored patients receiving continuous versus delayed DMF treatment, although clinical and neuroradiological improvements were observed following switch to DMF.
The safety profile observed in ENDORSE appears compatible with long-term use of DMF and is comparable with that of DEFINE/CONFIRM.3,4 Generally, similar incidences of AEs, AEs leading to discontinuation, and SAEs were observed among patients continuing DMF from the parent studies and those new to DMF.
Patients new to DMF had decreases in mean WBC and lymphocyte counts consistent with those of DMF-treated patients in DEFINE/CONFIRM. In patients continuing DMF, mean WBC and lymphocyte counts remained stable throughout time, and no further overall decrease in mean values was observed compared with the parent studies. In a post hoc analysis of DEFINE/CONFIRM; the clinical efficacy of DMF was not substantially different between patients with and without absolute lymphocyte counts less than the LLN. 26
Subsequent to the data cutoff for this report, a case of PML in a patient treated with DMF TID was reported in the setting of severe, prolonged lymphopenia. 10 Rare cases of PML also occurred in the post-marketing setting in the presence of prolonged lymphopenia (two cases in the presence of severe lymphopenia (approximately <500 mm3) and one case in the presence of moderate (nadir 600 mm3) lymphopenia, each persisting >6 months). Aside from these rare cases of PML, no overall increased risk of serious infections was noted, including other opportunistic infections. Analyses of data from phase 2 or phase 3 or ENDORSE studies support the importance of lymphocyte monitoring to identify patients experiencing lymphopenia persisting ⩾6 months. 26 Per recent labeling changes in the United States, a recent complete blood cell count (CBC), including lymphocytes, should be available before initiation of DMF, with retesting recommended after 6 months of treatment, every 6–12 months thereafter, and as clinically indicated. 11 Similarly, the European Medicines Agency (EMA) recommends obtaining a CBC before start of treatment and every 3 months during treatment. 27 Interruption of DMF should be considered for patients with lymphocyte counts <500/mm3 persisting for >6 months. Lymphocyte counts should be followed until recovery.
In ENDORSE, flushing, nasopharyngitis, and GI events were among the most commonly reported AEs. Flushing and GI events were more common among patients new to DMF in ENDORSE, particularly during the first year of treatment, consistent with the parent studies. 28 Although preliminary data on AE duration are reported, it should be noted that the studies were not designed to examine AE duration as an endpoint, particularly duration of episodic events, and there was a lack of temporal precision in reporting; results of the duration analyses should be interpreted cautiously. Phase 4 MANAGE study results of DMF in RRMS also indicate that GI-related events were generally transient and manageable. 29
The overall benefit–risk profile of DMF remains favorable. The sustained clinical and neuroradiological efficacy and AE profile observed in this interim analysis further support DMF as a valuable long-term treatment option for patients with RRMS.
Footnotes
Acknowledgements
Walter Rozdilsky, MS, from Complete Medical Communications (Hackensack, NJ, USA) wrote the first draft of the manuscript based on input from authors, and Elise Chahine from Complete Medical Communications (Hackensack, NJ, USA) copyedited and styled this article per journal requirements. Biogen reviewed and provided feedback on this article. The authors had full editorial control of this article and provided their final approval of all content. ENDORSE (Protocol 109MS303) is registered on www.clinicaltrials.gov (NCT00835770) and ![]() (EUDRA CT 2008-004753-14). The majority of the work on this article was conducted while M Novas and MT Sweetser were employees of Biogen. Statistical analyses were conducted by R Zhang, PhD; M Yang, MS; and J Potts, PhD, of Biogen, Cambridge, MA, USA. Drs Gold, Bar-Or, Hutchinson, Kappos, Havrdova, Selmaj, Fox and Phillips served on the medical advisory committee of parent study, contributed to the conception and design of the study, participated as an investigator and collected data, interpreted the data, and revised this article. Dr Arnold served on the medical advisory committee of parent study, contributed to the conception and design of the study, collected and interpreted data, and revised this article. Mr MacManus and Dr Yousry collected, analyzed, and interpreted the data and revised this article. Dr Pozzilli participated as an investigator and collected data, interpreted the data, and revised this article. Drs Sweetser and Novas contributed to the conception and design of the study, interpreted the data, and revised this article. Drs Zhang and Potts analyzed and interpreted the data and revised this article. Ms Yang served on the medical advisory committee of parent study, contributed to the conception and design of the study, analyzed the data, interpreted the data, and revised this article. Dr Miller contributed to the conception and design of the study, supported centralized collection of the magnetic resonance imaging (MRI) data, analyzed and interpreted the data, and revised this article. Dr Kurukulasuriya interpreted the data and revised this article.
(EUDRA CT 2008-004753-14). The majority of the work on this article was conducted while M Novas and MT Sweetser were employees of Biogen. Statistical analyses were conducted by R Zhang, PhD; M Yang, MS; and J Potts, PhD, of Biogen, Cambridge, MA, USA. Drs Gold, Bar-Or, Hutchinson, Kappos, Havrdova, Selmaj, Fox and Phillips served on the medical advisory committee of parent study, contributed to the conception and design of the study, participated as an investigator and collected data, interpreted the data, and revised this article. Dr Arnold served on the medical advisory committee of parent study, contributed to the conception and design of the study, collected and interpreted data, and revised this article. Mr MacManus and Dr Yousry collected, analyzed, and interpreted the data and revised this article. Dr Pozzilli participated as an investigator and collected data, interpreted the data, and revised this article. Drs Sweetser and Novas contributed to the conception and design of the study, interpreted the data, and revised this article. Drs Zhang and Potts analyzed and interpreted the data and revised this article. Ms Yang served on the medical advisory committee of parent study, contributed to the conception and design of the study, analyzed the data, interpreted the data, and revised this article. Dr Miller contributed to the conception and design of the study, supported centralized collection of the magnetic resonance imaging (MRI) data, analyzed and interpreted the data, and revised this article. Dr Kurukulasuriya interpreted the data and revised this article.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Gold has received consultant fees and grant or research support from Bayer HealthCare, Biogen, Genzyme, Merck Serono, Novartis, and Teva Neuroscience and compensation from Sage (editor fees). Dr Arnold has received revenue from Acorda Therapeutics, Biogen, EMD Serono, Genentech, Genzyme, GlaxoSmithKline, MedImmune, Mitsubishi, Novartis, Opexa Therapeutics, Receptos, Roche, Sanofi-Aventis, Teva, and NeuroRx Research and research support from Novartis and Biogen. Dr Bar-Or has received honoraria or research support from Biogen, DioGenix, Genentech, GlaxoSmithKline, Guthy-Jackson Charitable Foundation, Medimmune, Merck Serono, Novartis, Ono Pharmacia, Receptos, Roche, Sanofi-Aventis, and Teva Neuroscience. Dr Hutchinson has received honoraria from Bayer Schering, Biogen, Merck Serono, and Novartis and editorial fees from the Multiple Sclerosis Journal. Dr Kappos’ employer (University Hospital, Basel) has received steering committee or advisory board or consultant fees for Actelion, Addex, Bayer HealthCare, Biogen, Biotica, Genzyme, Lilly, Merck, Mitsubishi, Novartis, Ono Pharma, Pfizer, Receptos, Sanofi-Aventis, Santhera, Siemens, Teva, UCB, Xenoport; speaker fees for Bayer HealthCare, Biogen, Merck, Novartis, Sanofi-Aventis, Teva; educational support from Bayer HealthCare, Biogen, CSL Behring, Genzyme, Merck, Novartis, Sanofi, Teva; royalties from Neurostatus Systems GmbH; and grants from Bayer HealthCare, Biogen, Merck, Novartis, Roche, Swiss MS Society, Swiss National Research Foundation, European Union, and Roche Research Foundations. Dr Havrdova has received honoraria from Acetlion, Bayer, Biogen, Genzyme, Merck Serono, Novartis, Sanofi, Teva, Receptos, and Roche and support from Czech Ministry of Education, research project PRVOUK-P26/LF1/4. Mr. MacManus has received research grants from Apitope, Biogen, GlaxoSmithKline, Novartis, Richmond Pharma, and Schering AG. Dr Yousry has received research support from Biogen, GlaxoSmithKline, Novartis, and Schering AG; honoraria from Biogen, Bayer Schering, and Novartis. Dr Pozzilli has received consultant fees from Actelion, Biogen, Genzyme, Merck Serono, Novartis, and Teva Neuroscience and grant or research support from Biogen, Merck Serono, Novartis, and Teva Neuroscience. Dr Selmaj has received consultant fees from Genzyme, Novartis, Ono, Roche, Synthon, and Teva and fees for noncontinuing medical education (CME) services from Biogen. Dr Miller has received honoraria for advisory committee and/or consultancy advice in MS studies from Biogen, GlaxoSmithKline, Novartis, Merck, Chugai, Mitsubishi Pharma Europe, and Bayer Schering Pharma and compensation for performing central MRI analysis of MS trials from GlaxoSmithKline, Biogen, Novartis, and Apitope. Dr Fox has received consultant fees from Allozyne, Avanir, Biogen, Novartis, Questcor, Teva, and XenoPort and grant or research support from Novartis. Drs Zhang, Potts, Sweetser, Novas and Kurukulasuriya and Ms Yang are current or former employees of and hold stock or stock options in Biogen. Dr Phillips has received consultant fees from Acorda, Biogen, EMD Serono, Genzyme, and Sanofi and research support from Roche.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Biogen. Biogen provided funding for medical writing support in the development of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.