
Abstract
Background:
To date, no treatment for neuromyelitis optica (NMO) has been granted regulatory approval, and no controlled clinical studies have been reported.
Objective:
To design a placebo-controlled study in NMO that appropriately balances patient safety and clinical–scientific integrity.
Methods:
We assessed the “standard of care” for NMO to establish the ethical framework for a placebo-controlled trial. We implemented measures that balance the need for scientific robustness while mitigating the risks associated with a placebo-controlled study. The medical or scientific community, patient organizations, and regulatory authorities were engaged early in discussions on this placebo-controlled study, and their input contributed to the final study design.
Results:
The N-MOmentum study (NCT02200770) is a clinical trial that randomizes NMO patients to receive MEDI-551, a monoclonal antibody that depletes CD19+ B-cells, or placebo. The study design has received regulatory, ethical, clinical, and patient approval in over 100 clinical sites in more than 20 countries worldwide.
Conclusion:
The approach we took in the design of the N-MOmentum trial might serve as a roadmap for other rare severe diseases when there is no proven therapy and no established clinical development path.
Introduction
Neuromyelitis optica (NMO) is a rare, disabling autoimmune astrocytopathy typically characterized by severe and recurrent optic neuritis, longitudinally extensive transverse myelitis, and, less commonly, brain and brainstem lesions. Incomplete recovery from attacks is typical, giving rise to accumulative attack-related disability. NMO prevalence is estimated between 0.5 and 4.4/100,000.1,2 To date, no controlled clinical studies of NMO have been completed and no treatment has received regulatory approval. Immunosuppressive medications such as azathioprine, mycophenolate mofetil, mitoxantrone, prednisone, and rituximab are used empirically to prevent attacks, 3 although the evidence to support their use is limited. Robust clinical trials are required to demonstrate the safety and efficacy of potential new therapies for NMO.
One area of controversy within the NMO community and among regulatory agencies is the appropriateness of a placebo-controlled design in this disease.4–8 Although placebo control can provide unequivocal evidence of efficacy, there is legitimate concern about not treating patients since untreated subjects may be at higher risk of relapses (and their consequences) than those receiving empiric active treatments. Some investigators have argued that there is sufficient evidence to use empiric therapies as active comparators and that superiority to an active, rather than placebo, comparator is preferable. However, use of unproven active comparators could lead to results that are difficult to interpret. 9 For example, if the two active therapies appear equivalent, one would not know whether they are both effective or both ineffective (or even both harmful) as was the case in myasthenia gravis study comparing mycophenolate mofetil to prednisone. 10 If the study drug were found superior, it could be as a result of harm caused by the active comparator.
N-MOmentum (NCT02200770) is a clinical trial that randomizes NMO patients to receive MEDI-551, a monoclonal antibody that depletes CD19+ B-cells, or a placebo. The key considerations in the design of the N-MOmentum study included (1) establishing an ethical framework for conduct of a placebo-controlled trial in NMO, (2) assessing the “standard of care” for NMO, and (3) implementing study design features that provide a scientifically robust study and mitigate the risk associated with a placebo-controlled study.
Is there a “standard of care” for NMO?
A fundamental question is whether the immunosuppressive medications, currently used for NMO, constitute the “standard of care” and, if so, whether these medications need to be used as active controls in clinical trials of new medications. Strictly, to consider an intervention to be the standard of care, there should be robust scientific data demonstrating the effectiveness of the intervention for the treatment of the condition in question.
Regulatory agencies and providers may differ in the level of evidence required to consider an intervention “proven.” Regulatory agencies usually have a higher standard than those in clinical practice to make this determination. Nonetheless, some scientific criteria should be used to assess the available data and to determine whether a given treatment meets the standards of a proven therapy. The American Academy of Neurology (AAN) publishes Clinical Practice Guidelines for neurological interventions and therapies and categorizes them based on well-defined levels of evidence. 11 The AAN rates clinical studies as Class I–Class IV based on the scientific rigor of the study design. Well-controlled, multi-center, randomized trials are rated Class I. Single-center studies that are randomized are rated as Class II. Nonrandomized studies or ones in which historical controls are used are rated as Class III. Case series, case reports, and expert opinions are rated as Class IV. The recommendations for clinical practice are based on the class of the relevant studies and include four grades: “A,” “B,” “C,” and “U.” Level A recommendation requires at least two consistent Class I studies, while U is the lowest and is defined as data “inadequate” or “conflicting,” meaning that the treatment is “unproven.”
To objectively assess the current level of evidence of the use of immunosuppressive medications in NMO, a systematic literature review was performed using accepted methodology.12,13 A level of evidence was assigned to each study according to the AAN criteria described above. In total, 2438 citations were screened, and 105 studies (77 primary studies and 28 “kins,” defined as multiple publications of the same or overlapping series of patients) met inclusion criteria. An additional 116 case reports or case series with fewer than four patients were also identified. Of the 77 primary studies that met the inclusion criteria, 49 were studies of maintenance therapy to prevent NMO relapses. All studies that assessed current treatments to prevent NMO relapses were rated as Class IV evidence, and most were observational and retrospective in nature. This includes all published rituximab, azathioprine, steroid, mitoxantrone, and mycophenolate mofetil studies (list of references of these 49 studies is available upon request).
Typically, in the reviewed studies, the relapse rates in patients before and after a treatment is initiated were compared. Although many of these trials demonstrated an improvement in annualized relapse rate (ARR) or Expanded Disability Status Scale (EDSS) 14 score, the analysis is confounded by the absence of a contemporaneous control group, the variability in dosing regimens, limited reporting of safety data, the use of historical measures to gauge ARR prior to treatment, and other factors. For example, patients are often studied shortly after experiencing an attack, which may have exaggerated the baseline ARR; the post-treatment ARR could appear lower due to the well-recognized phenomenon of “regression towards the mean.” 15 These open-label observational studies rarely included sufficient methodological details to evaluate important confounding factors such as selection and information bias. Furthermore, the desire to identify a successful treatment may lead to biased reporting and evaluation of clinical data when the treatment assignment is not blinded. Based on this systematic review and applying the AAN treatment guidelines, all current treatments to prevent NMO relapses would be classified as “U” and therefore do not fulfill the criteria for establishing clinical guidelines or “standard of care” in the strict sense.
Although the European Federation of Neurological Societies (EFNS) guideline on management of NMO recommends immunosuppressive treatment, it also recognizes that there is “currently only class IV evidence for effect of any medication for relapse prevention” and that their recommendations are “based on expert opinion.” 16 Additional treatment guidelines published by expert consensus groups17,18 are also based on Class IV evidence. Whether one believes it is appropriate to have a placebo control in an NMO trial often depends on how strongly one believes that the currently used therapies have an unequivocally positive risk/benefit ratio. The available evidence is not strong enough to establish a standard of care that would prohibit a placebo-controlled trial in NMO.4,5 Once a treatment is established as effective and safe via a placebo-controlled, randomized trial, that treatment will become the standard of care, and a placebo control may not be needed or justified in subsequent NMO trials.
Ethical considerations for placebo-controlled studies in NMO
Similar to any other clinical investigation, placebo-controlled NMO treatment trials must respect patient autonomy, sufficiently balance risks and benefits, provide inherent value, and maintain justice. Of special interest in the NMO case are (1) the severity of the attacks, (2) the rarity of the condition, (3) the quality of evidence supporting use of current empirical treatments, and (4) disagreement within the clinical and scientific communities about the acceptability of a placebo arm in NMO clinical trials.
Clinical equipoise
Clinical equipoise requires (1) uncertainty (or honest professional disagreement in the community of experts) about the relative scientific and clinical merits of each of the treatment arms of the trial and (2) consistency of each treatment arm with competent medical care. The first of these requirements exists for NMO therapy; that is, we are uncertain whether and by how much the new treatment is better than no treatment or better than what is currently used empirically. The second requirement hinges on whether placebo can be considered competent medical care. This, in turn, depends on clinicians’ judgment of the risks of harm and the adequacy of the evidence of the effect of current empiric treatments.
Some assert that placebo treatment may represent a breach of a physician’s “duty of care.” If clinical equipoise exists, then a duty of care is not being breached. The decision regarding which comparator is ethically appropriate depends on (1) a value judgment about the adequacy of the evidence of the effect of the currently used treatments, (2) a value judgment about the relative (scientific and clinical) value or importance of answering the “placebo” question versus the (scientific and clinical) value of answering the “what we currently do” question, and (3) a value judgment that compares this scientific and clinical value against the relative risk of harm to those in the placebo control group. These judgments are complex and require substantial clinical and researcher expertise and patient input. They also require openness and an independent perspective.
Risk of harm
Given the potential severity of NMO relapses, risk of harm should be minimized for any clinical trial design, not only placebo-controlled trials. Several methodological strategies can be used to achieve this aim; however, none will remove the wrong of exposing some participants to some harm. The need for ethical judgment is not removed by minimizing overall harm to participants even if harm can be mitigated. That being said, no study design can eliminate all potential risks of harm. Harm may be caused by the disease under study or as an unintended consequence of treatment, for example, off-target effects. Part of the balance here rests on weighing the risks of harm associated with placebo (no treatment) versus the risks of harm associated with treatment. In the case of NMO, all current empiric therapies potentially cause harm including opportunistic infections and malignancies associated with immune suppressants. Potentially, the risks of current empiric treatments may be greater than placebo.
Consent
If the uncertainties associated with the available evidence for using current therapies along with the potential scientific and clinical values are openly and adequately explained to potential participants, their informed consent can be obtained. Even if these discussions are difficult, complex, or differ from what has previously been discussed with the patient, informed consent is possible.
Rarity and severity
Finally, the rarity and the severity of NMO make a difference in the ethics of conducting research in this area. Since NMO is rare, we may think that the sufferers should be more protected, or we may think we need to go to greater lengths to ensure that this research is conducted so that future suffering can be avoided. In the case of severity, we are hesitant to expose patients with severe conditions to potential risk, yet the severity of the disease makes the importance of finding effective treatments even greater. These competing issues raise the stakes in NMO clinical research and polarize the positions taken by stakeholders in this clinical research area. Therefore, even more we need to rely on the processes that we use to make these judgments, and this require openness, independence, and inclusiveness.
Features of the N-MOmentum study that will reduce the potential risk of placebo
Given the issues discussed above, the following design features were incorporated to mitigate the potential risk associated with a placebo-controlled study (Table 1).
Challenges faced in the design of the N-MOmentum trial and the solutions that were implemented.
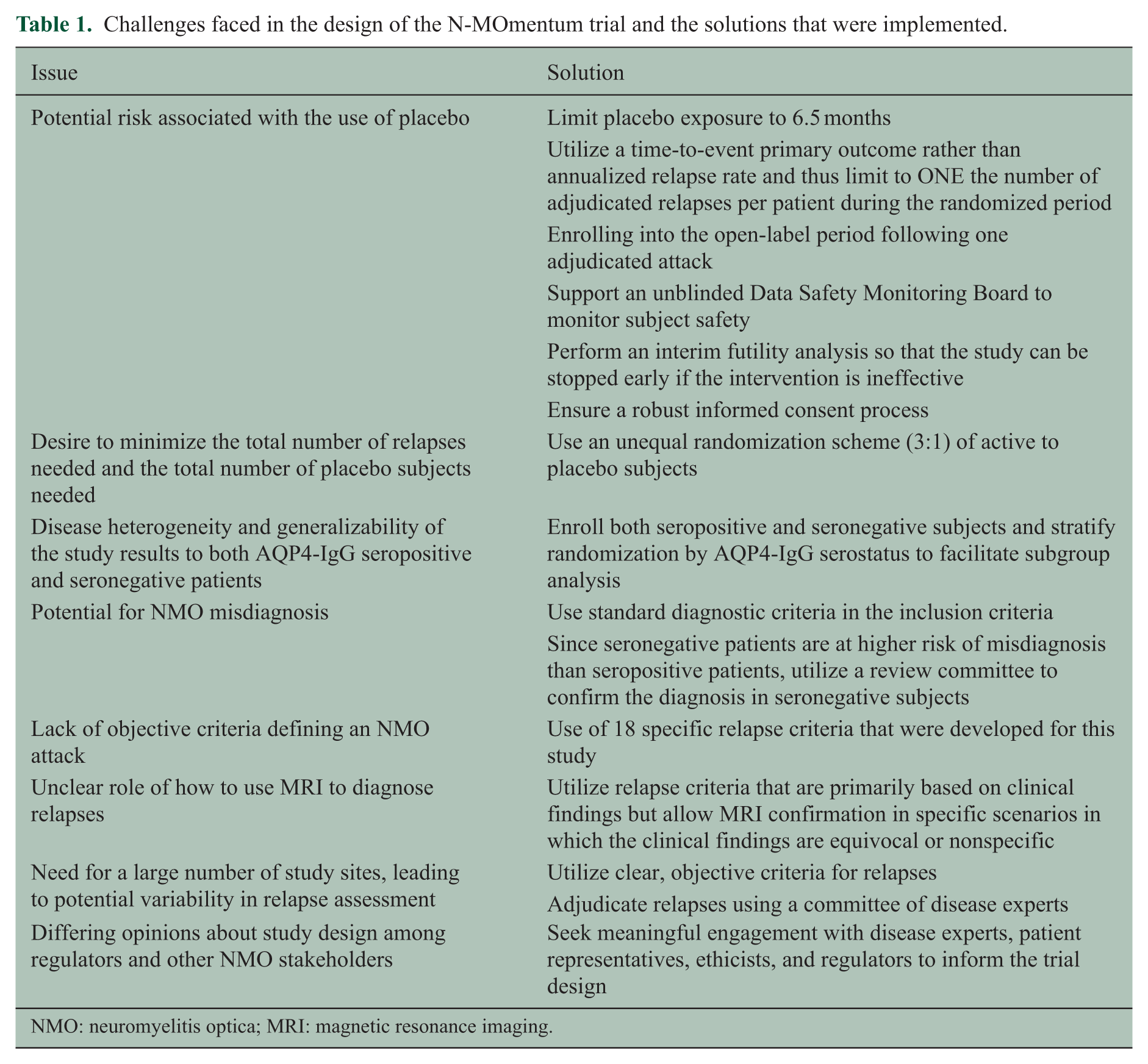
NMO: neuromyelitis optica; MRI: magnetic resonance imaging.
Time-to-attack primary outcome
Relapses are the most medically relevant biological event in NMO, and proof of efficacy can be based on delaying and/or reducing the occurrence of relapses. Given the potential severity of NMO attacks, exposing placebo-treated patients to multiple attacks during the randomized portion of the study was deemed to be unacceptable. In clinical practice, many neurologists initiate or switch treatment following an NMO attack. Therefore, the primary endpoint of this study is time-to-attack within the masked, randomized, placebo-controlled part of the study. This ensures that patients in this study and especially those on placebo will not experience more than one attack while on study drug. This endpoint has been used effectively in several clinical trials such as clinically isolated syndromes suggestive of initial demyelinating events associated with multiple sclerosis (MS).19–22 Patients who experience an attack are eligible for open-label treatment with MEDI-551, following investigator-determined rescue therapy. In the open-label period, patients will receive MEDI-551 every 6 months until the study is terminated or, if successful, until regulatory approval.
Limited duration of potential placebo exposure
Another study design element that reduces the risk of placebo exposure is limiting the duration of the randomized controlled period to 6.5 months. Randomized controlled trials in MS have used a 6-month duration for proof-of-concept Phase II studies. Several placebo-controlled clinical trials in MS have shown a significant impact of treatment on MS relapse frequency over a 6-month time frame (natalizumab, 23 fingolimod, 24 rituximab, 25 and ocrelizumab 26 ). Given that the goal of this study is to show an impact on time to first relapse and that the frequency of relapses in NMO is expected to be higher than in MS, limiting the study to 6.5 months of placebo exposure will probably be sufficient to detect a clinically important treatment effect and will enhance subject recruitment. Subjects who experience an adjudicated relapse or who complete the 6.5-month randomized controlled period without an adjudicated relapse will be offered participation in the open-label period.
Unequal randomization
Our estimate of the effect size of MEDI-551 is based on nonrandomized studies of rituximab. It is to be expected that MEDI-551 will share the same class effects that have been seen with other B-cell depleting agents in patients with NMO. Feedback from investigators and patients indicated that 3:1 randomization would be easier to accept for a placebo-controlled design as opposed to 2:1 or 1:1. One downside of unequal randomization is that it increases the total number of patients needed in the trial, but not as much as an active agent comparative trial. However, the 3:1 ratio requires fewer placebo patients (53, compared to 56 and 65 in 2:1 and 1:1 ratios, respectively), and fewer attacks are estimated under the alternative hypothesis in the placebo group (27, compared to 30 and 34 in 2:1 and 1:1 ratios, respectively).
Unblinded Data Safety Monitoring Board and interim futility analysis
Safety and efficacy data from the trial will be reviewed periodically by an unblinded Data Safety Monitoring Board (DSMB). They will be able to advise the study sponsor if any safety concerns are identified during the conduct of the trial. The DSMB will also perform an unblinded futility analysis after 34 attacks (~50% of the total expected attacks in the two study groups combined) occur. This will enable an early determination of whether the study is futile, thus potentially reducing the number of subjects who are exposed to an ineffective treatment.
Other considerations in the design of the N-MOmentum trial
Definition of the study population
Misdiagnosis of NMO as MS or other diseases is a clinical challenge that needs to be addressed in the design of an NMO clinical trial. Fortunately, the anti-AQP4 antibody (AQP4-IgG) is highly specific for NMO/NMO spectrum disorders (NMOSD) and thus is very useful as a diagnostic biomarker. Hence, the main concern for potential misdiagnosis is for the seronegative patients. To increase the homogeneity of the study population, some trials have excluded seronegative patients. The disadvantage of this strategy is that the trial will exclude a cohort of patients who meet established diagnostic criteria 27 and fail to establish clinical efficacy in this disease cohort. Along with seropositive patients, the N-MOmentum trial will enroll seronegative subjects who meet diagnostic criteria for NMO 27 (20% of subjects enrolled). To ensure that seronegative patients meet diagnostic criteria for NMO, a three-member independent Eligibility Committee will review all relevant data and confirms the diagnosis. The sample size determination for this study assumed that the proportion of AQP4-IgG seropositive cases is 80%. This strategy is well aligned with the principles outlined in US Food and Drug Administration (FDA) 28 guidance on enrichment for definite cases, but not excluding cases without a biomarker, when there is a reasonable expectation that both those with and without the diagnostic biomarker may respond similarly.
Subjects will need to have experienced at least one clinical attack in the last year or two clinical attacks in the last 2 years. The age range for the trial is broad (18 years and above), and subjects are permitted to enter with significant neurological disability as reflected by EDSS range up to 8.0. Treatment with mycophenolate mofetil, azathioprine, or daily prednisone is allowed up to the point of randomization. Rituximab-treated subjects will also be allowed at entry; however, these subjects will need to have B-cell repletion to at least the lower limit of normal. These entry criteria will allow the participation of many NMO subjects, and results of the study could be generalizable to the majority of the NMO population.
Definition and adjudication of NMO attacks
As the primary outcome, it is critical that attacks are rigorously defined and specific criteria are implemented to identify specific NMO-related events as early as possible. Because there are no evidence-based definitions of NMO attacks, a panel of NMO experts defined 18 NMO attack criteria: 11 for optic neuritis, 4 for myelitis, 2 for brainstem, and 1 for hemispheric involvement (Table 2). These criteria are primarily clinical but utilize magnetic resonance imaging (MRI) for confirmation in cases where the clinical findings are equivocal or nonspecific. The US FDA accepted these criteria.
NMO attack criteria.

CF: counting fingers; EDSS: Expanded Disability Severity Score; FSS: Functional System Scores; Gd: gadolinium; HM: hand motion; LP: light perception; MRI: magnetic resonance imaging; NLP: no light perception; NMO/NMOSD: neuromyelitis optica/neuromyelitis optica spectrum disorders; ON: optic neuritis; RAPD: relative afferent pupillary defect.
The symptoms listed are examples and are not inclusive of all NMO/NMOSD symptoms.
Four major areas of the body may be affected by an attack: the optic nerve, resulting in ON; the spinal cord, resulting in myelitis; the brainstem, resulting in a number of outcomes; and the brain.
Symptom(s) must meet at least one of the objective criteria for an NMO/NMOSD attack. However, the symptom(s) may meet more than one of the criteria for an NMO/NMOSD attack across different body systems.
At least 2-step drop can be any of the following worsening: on Landolt C Broken Rings Chart to HM, LP, or NLP; CF to LP or NLP; and HM to NLP.
At least 1-step drop can be any of the following worsening: on Landolt C Broken Rings Chart to CF, HM, LP, or NLP; CF to HM or LP or NLP; and LP to NLP.
A 1-point change in a single FSS without a change in the EDSS, with or without a new Gd-enhancing or new/enlarging T2 MRI lesion in the spinal cord, is not considered a clinically significant change and will not count as an attack per this protocol.
Three general types of NMO attacks have been described: (1) myelitis, (2) optic neuritis, and (3) brainstem or cerebral attacks. Myelitis attacks are based on a change in the pyramidal, sensory, or bowel and bladder Functional System (FS) scores of the EDSS that would be affected by this type of attack. In patients with clinically significant ambulatory impairment, a change in the EDSS score can be used to define an attack. In milder cases of myelitis, confirmation requires identification of a new, enlarging or active (gadolinium-enhancing) MRI lesion in the spinal cord. Optic neuritis attacks are based on visual acuity changes and presence of a new relative afferent pupillary defect (RAPD). In milder cases of optic neuritis, confirmation requires documentation of a new, enlarging or active (gadolinium-enhancing) MRI lesion in the anterior visual pathway. For example, a subject with a 10-character drop in high-contrast visual acuity and a new RAPD during an optic neuritis event would not need MRI confirmation. In contrast, a patient with a 5-character drop and no new RAPD would need to also have a new MRI lesion in the corresponding optic nerve to meet relapse criteria. Another example of the need for MRI-supported relapse criteria is the most common brainstem attack in NMO: a lesion in the area postrema that manifests with nausea, vomiting, or hiccups. When persistent, these symptoms fulfill the clinical requirements for a NMO relapse; however, given the lack of specificity of these symptoms, identification of an area postrema lesion on MRI is required to confirm inflammatory injury. For attacks that involve the cerebral hemispheres, changes in relevant FS subscores can be used to define the relapse in conjunction with identification of an appropriately located new or active MRI brain lesion.
Given the rarity of this disease, the number of participating sites will be high and the number of patients enrolled at each site few. To minimize variability amongst the sites in determining the occurrence of NMO attacks, a three-member masked Adjudication Committee will review all attack assessments within 14 days of attack assessment visit initiation. The committee will review clinical data, and when clinical criteria require MRI confirmation, MRI data. Only adjudicated attacks will count toward the primary endpoint and allow subjects to enter into the open-label period. However, the adjudication process will not influence the site investigator’s decision regarding relapse treatment.
Maintenance of blinding
Because the behavior of patients, investigators, and assessors might be biased by beliefs about treatment allocation, it is critical to maintain blinding to treatment allocation. The 3:1 randomization could lead site staff to assume (sometimes incorrectly) that the patient is on the active drug and thereby downplay relapse risk. However, the concern about the placebo may cause relapse over-diagnosis. The double-masked design of the study is aimed at mitigating this risk. To further reduce the risk of bias, the study includes defined clinical criteria to assess the NMO attacks, masked EDSS and visual acuity assessors, and a central attack adjudication committee.
Statistical considerations
There were several challenges to determining the expected frequency of relapses in NMO in order to calculate the trial sample size. First, because large-scale natural history studies in NMO have not been done, it was necessary to use a meta-analytic approach to evaluate recent literature. Second, evolving NMO diagnostic criteria can affect relapse rate estimates using different NMO patient populations. Therefore, our analysis included studies that used the published diagnostic criteria for NMO that were in use at that time. 27
Meta-analysis of recent literature led to estimates of the hazard rate for the placebo group to be 1.5 and 1.0 relapses per year for seropositive and seronegative, respectively, based on the pre-treatment relapse frequency.29–33 The target treatment effect (reduction in risk of attack) with MEDI-551 was estimated to be 60% based on another meta-analysis of open-label azathioprine 30 and rituximab studies.29,31,32 Thus, with 3:1 randomization, target hazard ratio of 0.4, hazard rates in the placebo-treated group quoted above, and Type I error (two-sided) of 5%, the number of events needed is estimated as 67 to achieve 90% power to show a statistically significant difference between active treatment and placebo. Assuming 28 weeks of controlled randomized period, we predicted that the required sample will be approximately 212 subjects. If the actual on-study annual relapse hazard rate is less than expected, a larger number of subjects will be needed for the 67 relapses to occur within the 28-week randomized, controlled period. Randomization will be stratified by seropositivity to AQP4-IgG and by whether or not the patient is Japanese (to enable analysis of this subpopulation). Figure 1 outlines the study design schematically.
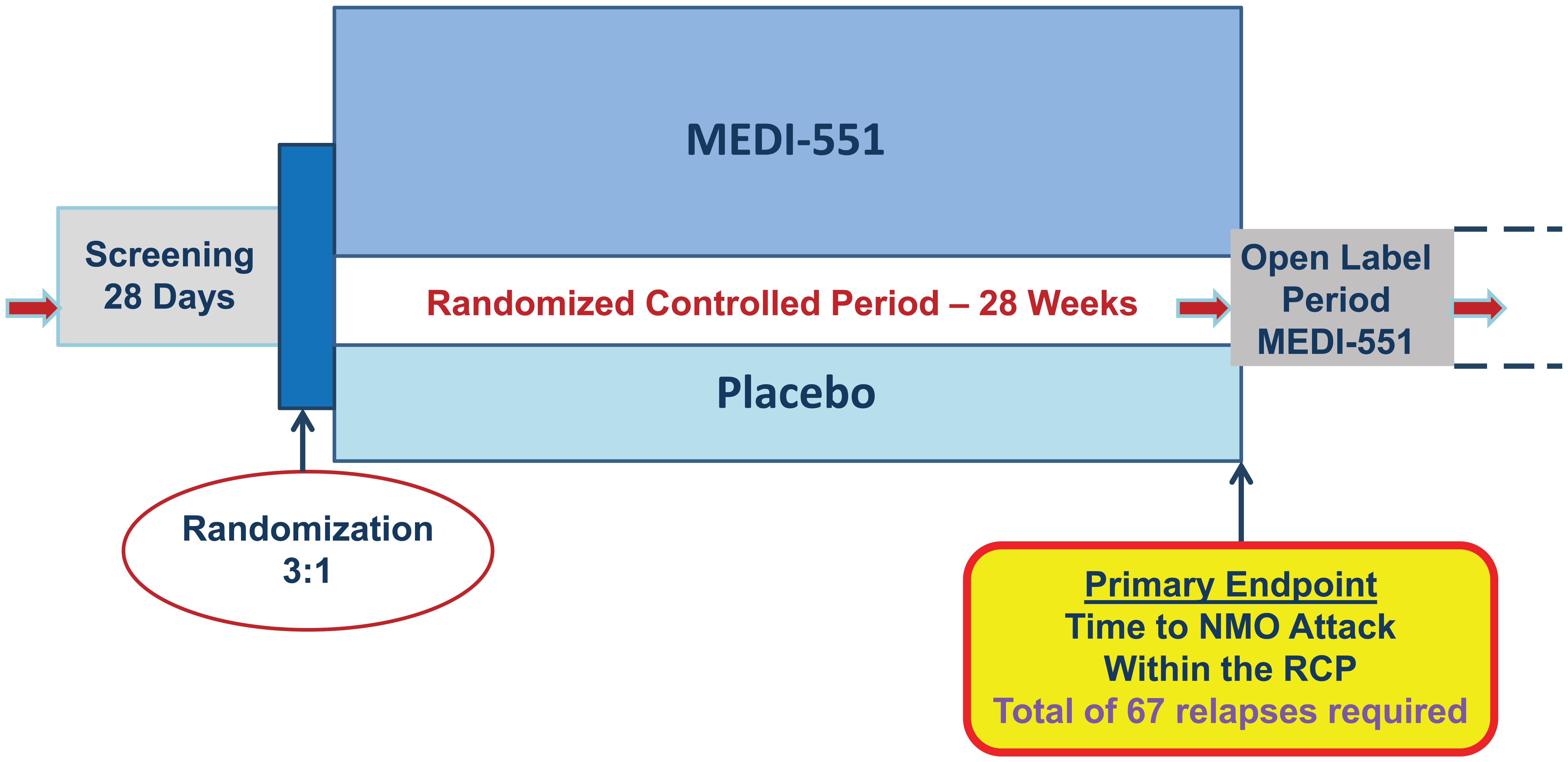
N-MOmentum study design scheme.
Regulatory and community perspective
Key to the success of any clinical development program is regulatory acceptance. Like the medical community as a whole, regulatory agencies have provided a spectrum of opinions on the optimal trial design with regard to use of a placebo control. Following public discussion as well as direct exchange of opinion between the study sponsor and the regulatory agencies, most regulatory authorities around the world have accepted the N-MOmentum study design.
As of 31 August 2015, 118 sites have been identified and agreed to participate. Numerous letters of support were received from principal investigators and key opinion leaders in NMO from around the world. In all, 21 countries’ regulatory authorities and country-level ethics committees/institutional review boards (IRBs) have approved the study. More importantly, as of the time of writing, 57 patients from different parts of the world have signed an informed consent and entered the study; of these, 21 have been randomized into the two treatment arms of the study.
Conclusion
NMO poses a number of development challenges because it is a rare, severe disease with no proven therapies and is currently treated empirically. Since registration studies have not been conducted in NMO, agreed endpoints and study designs are additional challenges. In this setting of a rare disease with unequivocal unmet need, conducting the most robust trial to provide interpretable data is critical. We described the status of current treatments and the ethical considerations that led to the N-MOmentum clinical trial that is specifically designed to mitigate the risks of placebo control while maintaining scientific rigor.
The medical/scientific community, patient organizations, and regulatory authorities were engaged in early discussions on this study, and their input contributed to the final study design. As a result, the N-MOmentum study design has garnered global regulatory, ethical, clinical, and patient approval. Similar considerations should be addressed in advance for any rare disease with no approved medications.
Footnotes
Acknowledgements
The authors would like to express their deep appreciation to the patients and investigators whose participation makes this study a reality. The Authors would like to thank Peloton Advantage for the editorial support funded by MedImmune.
Conflict of interest
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: O. Aktas has served as a consultant for MedImmune and as a speaker for Bayer, Biogen, Genzyme, Merck Serono, Novartis, and Teva, and his institution has received research funding and grants from Bayer, Biogen, the German Research Foundation, and Novartis. JL Bennett holds patents for compositions, including blocking monoclonal therapy, and treatment methods for neuromyelitis optica; has served as a consultant for AbbVie, Alnylam Pharmaceuticals, Chugai Pharma, EMD Serono, Genentech, Genzyme, MedImmune, and Novartis; has received research support from the Guth-Jackson Foundation, the NIH, Novartis, and Questcor Pharmaceuticals; holds stock in Apsara Therapeutics; and receives royalties from Aquaporumab. J. Cohen has served as a consultant for Novartis and his institution has received research support from Genzyme, Novartis, Receptos, Synthon, and Teva. B. Cree has served as a consultant and has developed educational presentations for MedImmune. G. Cutter has served as a consultant, speaker, and advisory board member for the Consortium of MS Centers, D3 (Drug Discovery and Development), EMD Serono, Genzyme, Genentech, Jannsen Pharma-ceuticals, Klein-Buendel Incorporated, MedImmune, Novartis, Opexa Therapeutics, Receptos, Roche, Spiniflex Pharmaceuticals, Somahlution, Teva, and Transparency Life Sciences; he has also participated on Data and Safety Monitoring Boards for Apotek, Biogen-Idec, Cleveland Clinic, GlaxoSmithKline, Gilead, Horizon Pharmaceuticals, Modigenetech/Prolor, Merck/Ono Pharmaceuticals, Merck, Merck/Pfizer, the National Heart, Lung, and Blood Institute, the National Institute of Neurological Disorders and Stroke, the National Institute of Child Health and Human Development, Neuren, Sanofi-Aventis, and Teva. K. Fujihara has served as a consultant and developed educational presentations for MedImmune, and his institution has received research support and payment for lectures from MedImmune. H-P Hartung has served as a consultant for Apexa, Biogen GmbH, GeNeuro, Merck Serono, Novartis, and Octapharma. HJ Kim has served as a consultant, speaker, and advisory board member for MedImmune, and has received research funding and paid travel and accommodations from MedImmune. F. Paul has received research funding and grants from various pharmaceutical companies, government agencies, and private bodies; serves on the editorial board of Neurology; and his institution has received consultancy fees from Alexion Pharmaceuticals, MedImmune, and Novartis. S. Pittock has served as a consultant for Alexion Pharmaceuticals, Chugai Pharma, and MedImmune; has received a research grant from the National Institutes of Health and research funding from Alexion Pharmaceuticals; and holds several patents. M. Sheehan’s institution has received consultancy fees and research funding from MedImmune. B. Weinshenker has served as a consultant for Chord Therapeutics, Chugai Pharma, and Novartis; received a grant from the Guthy Jackson Charitable Foundation; holds a patent with Mayo Medical Ventures and receives royalties from RSR and Oxford University; has received paid travel and accommodations from Alexion Pharmaceuticals; has participated on Data and Safety Monitoring Boards for Biogen-Idec, Mitsubishi, and Novartis; and has served on an adjudication committee for a clinical trial for MedImmune. D. Wingerchuk’s institution has received consultancy fees from Alexion Pharmaceuticals and MedImmune and has received research funding from Alexion Pharmaceuticals and Terumo BCT. E. Katz, S Madani, G. Barron, A. Flor, J. Ratchford, and K. Patra are employees of MedImmune and own stock in AstraZeneca. No author received an honorarium or other form of payment for the preparation of this manuscript.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.