
Abstract
Objective:
The purpose of this study was to determine the effects of oral teriflunomide on multiple sclerosis (MS) pathology inferred by magnetic resonance imaging (MRI).
Methods:
Patients (n=1088) with relapsing MS were randomized to once-daily teriflunomide 7 mg or 14 mg, or placebo, for 108 weeks. MRI was recorded at baseline, 24, 48, 72 and 108 weeks. Annualized relapse rate and confirmed progression of disability (sustained ≥12 weeks) were the primary and key secondary outcomes. The principal MRI outcome was change in total lesion volume.
Results:
After 108 weeks, increase in total lesion volume was 67.4% (p=0.0003) and 39.4% (p=0.0317) lower in the 14 and 7 mg dose groups versus placebo. Other measures favoring teriflunomide were accumulated enhanced lesions, combined unique activity, T2-hyperintense and T1-hypointense component lesion volumes, white matter volume, and a composite MRI score; all were significant for teriflunomide 14 mg and most significant for 7 mg versus placebo.
Conclusions:
Teriflunomide provided benefits on brain MRI activity across multiple measures, with a dose effect evident on several markers. These effects were also consistent across selected subgroups of the study population. These findings complement clinical data showing significant teriflunomide-related reductions in relapse rate and disease progression, and demonstrate containment of MRI-defined disease progression.
Keywords
Introduction
Teriflunomide is a novel, oral, disease-modifying therapy (DMT) recently approved in the USA and Australia, and currently under review by the European Medicines Agency for the treatment of relapsing forms of multiple sclerosis (RMS). Teriflunomide selectively and reversibly inhibits dihydro-orotate dehydrogenase, a key mitochondrial enzyme in de novo pyrimidine synthesis required by rapidly dividing lymphocytes. Through this cytostatic effect, teriflunomide limits expansion of stimulated T and B cells thought to be responsible for the damaging inflammatory process associated with MS. Slowly dividing or resting cells, including lymphocytes and non-lymphoid cells, rely on the pyrimidine salvage pathway to meet their pyrimidine demand. As the pyrimidine salvage pathway is unaffected by teriflunomide, basic homeostatic functions of resting and slowly dividing cells appear to be preserved, and lymphocytes remain available for immune surveillance. 1,2
A phase II study demonstrated that teriflunomide (7 or 14 mg/day) reduced magnetic resonance imaging (MRI) activity measures by over 61% versus placebo and was well tolerated over 36 weeks. 3 This trial’s ongoing open-label extension showed that these benefits were sustained over eight years. 4 In addition, phase II studies that evaluated the addition of teriflunomide to patients with RMS already receiving stable treatment with interferon beta 5 or glatiramer acetate 6 showed no unique safety concerns for either combination with MRI evidence of additive effects.
The Teriflunomide Multiple Sclerosis Oral (TEMSO) trial (ClinicalTrials.gov: NCT00134563) is the first published phase III study from the clinical development program. 7 Teriflunomide 7 mg or 14 mg/day significantly reduced the annualized relapse rate (ARR) by over 30% (p<0.001), and the 14 mg dose reduced the risk of 12-week confirmed disability progression by 30% (p=0.03) compared with placebo. 7
MRI is frequently used to evaluate the efficacy of DMTs in MS trials. Common measures include change in number and volume of enhanced lesions seen on T1-weighted images following administration of gadolinium (Gd) chelates, number and volume of hyperintense lesions found on T2-weighted or fluid attenuation by inversion recovery (FLAIR) images, number and volume of hypointense lesions on T1-weighted images, and estimates of brain parenchymal tissue loss from baseline to predefined trial intervals. Enhanced lesions (Gd+) indicate blood–brain barrier breakdown marking regions with acute inflammation. An important activity measure is the number of unique active lesions (UALs; Gd+ lesions plus unenhanced new and substantially enlarged T2-hyperintense lesions) identified on relatively infrequent sequential imaging. 3 Measures of UALs correlate with clinical relapse rates; consequently, the effects of DMTs on lesion activity correlate with their effects on clinical relapses 8 and accumulated disability within the context of clinical trials. 9 T2 lesion volume provides some measure of past disease activity, while T1-hypointense lesions indicate more severe damage reflecting axonal loss, gliosis and loss of intracellular matrix. 10 Measures such as reduction in brain volume are an additional indication of disease progression. 11
The primary clinical and key MRI efficacy outcomes of TEMSO are already published; 7 here we report secondary and exploratory MRI outcomes.
Methods
Patient population and study design
The TEMSO clinical trial design and overall outcomes have been published elsewhere. 7 In brief, eligible patients were aged 18–55 years, met McDonald’s diagnostic criteria, 12 exhibited a relapsing course with or without progression, had a Kurtzke Expanded Disability Status Scale (EDSS) score ≤5.5 and experienced at least two clinical relapses in the prior two years or one relapse during the preceding year, but no relapses within 60 days of randomization. Following consent and screening, eligible patients were randomized. MRI scans were performed at baseline and weeks 24, 48, 72, and 108. Participants completing the study could enter a long-term extension.
Study endpoints
The key clinical efficacy outcomes of TEMSO were ARR (primary endpoint) and disability progression as assessed by changes in the EDSS score (key secondary endpoint). The key MRI endpoint was the change in total lesion volume from baseline, defined as the total volume of the T2-hyperintense and T1-hypointense lesion components. Secondary MRI outcomes included the number and volume of enhancing lesions per scan, and the volume of T2-hyperintense and T1-hypointense lesion components. Pre-specified exploratory variables included the proportion of patients free from Gd+ lesions, the number of UALs per scan, Z4 composite score, 13 and change in brain parenchymal fraction (BPF; defined as change in the fraction of segmented intracranial contents not classified as cerebrospinal fluid (CSF) divided by the total segmented intracranial contents), gray matter (GM) and white matter (WM) volumes.
MRI procedures
MRI of the brain with and without contrast was acquired using a standardized protocol with seven scan series. MRI site certification was completed prior to activation of the clinical sites with recertification scans required for machine changes or major upgrades. Scanners were restricted to General Electric, Philips, and Siemens machines of 1.5 (123 MRI centers) or 3.0 (three MRI centers contributing a total of four subjects) Tesla field strengths; one subject was shifted to a 3T imager during the trial. Baseline MRI was received at least one week before randomization; subsequent scans were performed within seven days of scheduled visits. Semi-automated processing was used to extract the various tissue volumes. Serial registered sets of fully processed dual fast spin echo, FLAIR, pre- and post-Gd T1, final segmented and Gd-seeded images underwent expert review to enumerate new or substantially enlarged T2-hyperintense lesions that lacked enhancement (see Supplementary Material for detailed methodology).
Statistical analysis
Change from baseline in total lesion volume was analyzed using a mixed-effect model with repeated measures (MMRM) on cubic root transformed volume data. 14 The model included factors (fixed effects) for treatment, EDSS strata, region, visit, treatment-by-visit interaction, baseline lesion volume and baseline-by-visit interaction. The same type of analysis was applied to the change from baseline in T1-hypointense lesion volume and the strictly T2-hyperintense lesion component. Changes in the volume of GM and WM, BPF and Z4 scores were analyzed using MMRM with the model factors included. Z4 scores were constructed based on the addition of z-transformation of the Gd+ tissue volume, total lesion volume, T1-hypointense lesion volume and normalized CSF volume (1-BPF). 13,15
The numbers of enhanced lesions and UALs per MRI scan were compared among groups using a Poisson regression model with robust error variance. The model included total lesion numbers as response variables, and treatment group, EDSS strata, region, and either baseline number of enhanced lesions or UALs as covariates, and log- transformed number of scans as an offset variable. The total volume of enhanced lesions per MRI scan was analyzed using rank analysis of covariance. The adjustment for covariance in the rank and strata used an analysis of covariance model that included the ranked volume of enhanced lesions per scan as the response variable and ranked baseline volume of enhanced lesions, EDSS strata and region as covariates.
Post-hoc sensitivity analyses were performed for change in total lesion volume, and numbers of on-study enhancements and UALs based on patients’ entry characteristics that included age (split on the median of <38 or ≥38 years), sex, entry EDSS strata (≤3.5 or >3.5), number of relapses within two years prior to randomization (0–1, 2, 3, or ≥4), MS subtype (relapsing–remitting, secondary progressive, or progressive relapsing), prior use of other DMTs and baseline MRI parameters (presence or absence of enhanced lesions; total lesion volume (split on the median of <13 or ≥13 ml). All other endpoints were analyzed using the same statistical methods as for the primary analysis described above.
Results
Between September 2004 and March 2008, 1088 patients from 126 centers were randomized. Fourteen patients were excluded from the MRI analyses based on missing baseline data, leaving 1074 evaluable cases: 359 in the placebo, 359 in the 7 mg, and 356 in the 14 mg teriflunomide groups. No significant differences between treatment groups were observed in baseline demographics (Table 1).
Patient demographics and magnetic resonance imaging (MRI) characteristics at baseline.
SD: standard deviation. All volumes are given in ml.
Key MRI variable
The previously reported key MRI outcome, change in total lesion volume from baseline, was significantly lower for patients randomized to active treatment. 7 At week 108, the least squares (LS) mean difference from placebo in transformed total lesion volume was −0.053 (95% confidence interval (CI): −0.101 to −0.005; p=0.0317) for the 7 mg and −0.089 (95% CI: −0.137 to −0.041; p=0.0003) for the 14 mg teriflunomide groups. The mean (standard deviation, SD) change in total lesion volume from baseline was 2.21±7.00, 1.31±6.80 and 0.72±7.59 ml, for the placebo, 7 mg and 14 mg teriflunomide arms, respectively. The respective median changes were 1.127, 0.755 and 0.345.
Secondary MRI variables
Number of Gd+ lesions
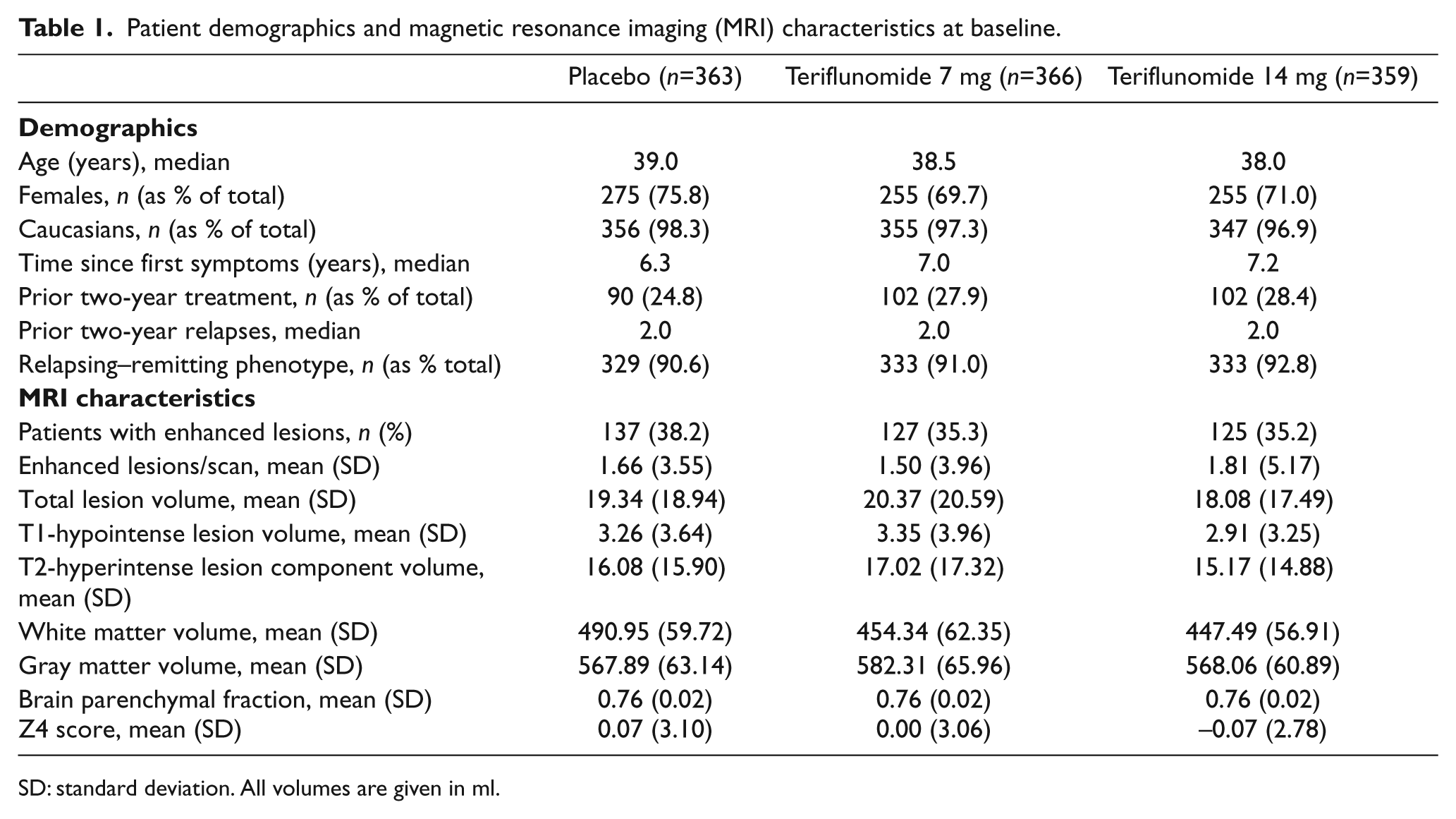
The cumulative number of enhancements from baseline in the three arms of the trial is shown in Figure 1. The adjusted number of Gd+ lesions per scan at 108 weeks was 1.331 (95% CI: 1.059 to 1.673) in the placebo group compared with 0.570 (95% CI: 0.434 to 0.748) and 0.261 (95% CI: 0.167 to 0.407) in the 7 mg and 14 mg groups, respectively. This represents relative risk reductions of 57.2% (p<0.0001) in the 7 mg and 80.4% (p<0.0001) in the 14 mg groups compared with placebo. A dose response was also apparent in the proportion of patients free from enhanced lesions: 51.4% in the 7 mg group and 64.1% in the 14 mg group, compared with 39% of the placebo group. Post-hoc analyses of the difference between the 7 mg and 14 mg doses of teriflunomide on the number of enhanced lesions per scan and proportion of Gd+ lesion-free patients were both significant (p=0.0024 and p<0.001).
Cumulative number of enhancements from baseline.
T1-hypointense lesion volume
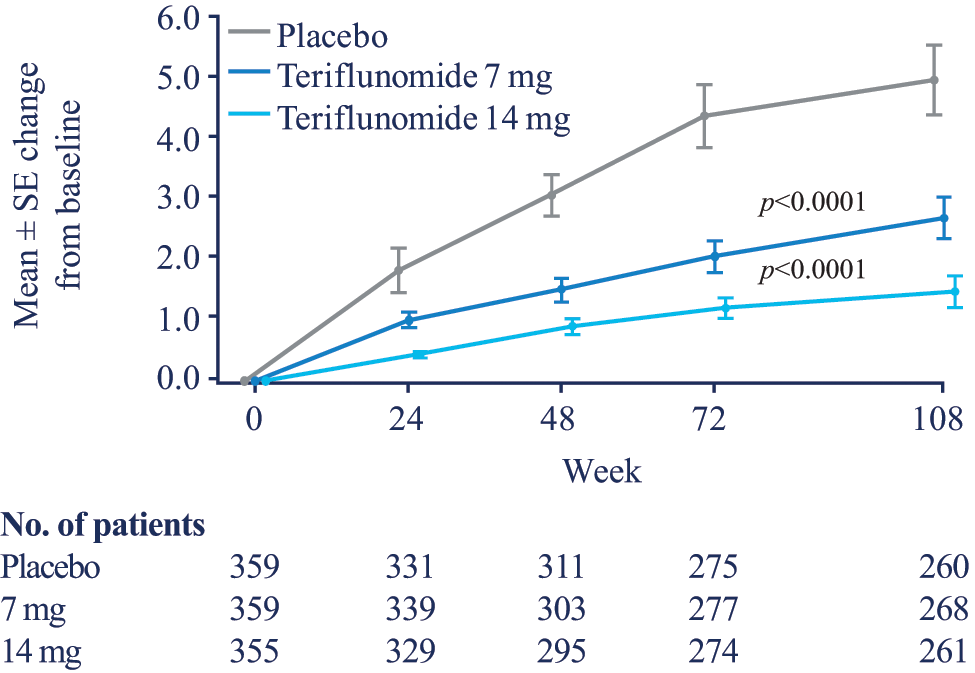
At week 108, the LS mean difference from placebo in transformed T1-hypointense lesion volume was −0.016 (95% CI: −0.041 to 0.008; p=0.1916) for the 7 mg and −0.030 (95% CI: −0.055 to −0.006; p=0.0161) for the 14 mg groups. For the teriflunomide 14 mg group, this was a 31.3% relative change from placebo. The change in the volume of the T1-hypointense lesion component over time is shown in Figure 2. Significant differences from placebo were evident at all intervals after baseline for both actively treated groups through to week 72 of the study, but were maintained through to week 108 only in the 14 mg group.
Change from baseline in the T1-hypointense lesion volume component.
T2-hyperintense lesion component volume
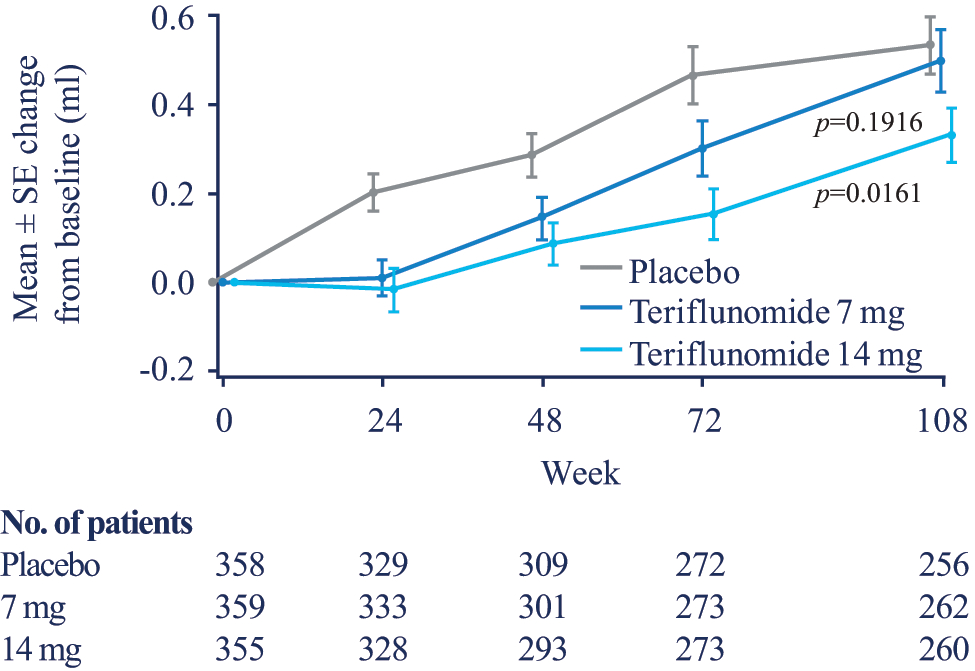
The T2-hyperintense component lesion volume was reduced over time in both teriflunomide groups compared with placebo, with a marked decrease for the 14 mg group statistically evident in the earliest MRI scans at 24 weeks. The treatment reduction for the 7 mg group was marginal at week 24 (p=0.0523), but well established at week 48 (Figure 3). At week 108, the LS mean difference from placebo in transformed T2-hyperintense lesion component volume was −0.051 (95% CI: −0.100 to −0.002; p=0.0404) for the teriflunomide 7 mg group and −0.089 (95% CI: −0.139 to −0.040; p=0.0004) for the 14 mg group. The T2-hyperintense lesion component was reduced by 44.0% and 76.7% in the 7 mg and 14 mg groups, respectively.
Change from baseline in the strictly T2-hyperintense lesion component volume.
Exploratory MRI outcomes
UALs
The number of UALs per scan at the end of treatment was significantly lower in both teriflunomide treatment groups, corresponding to a relative risk reduction of 47.7% (p<0.0001) in the 7 mg and 69.4% (p<0.0001) in the 14 mg groups, compared with placebo (Figure 4).
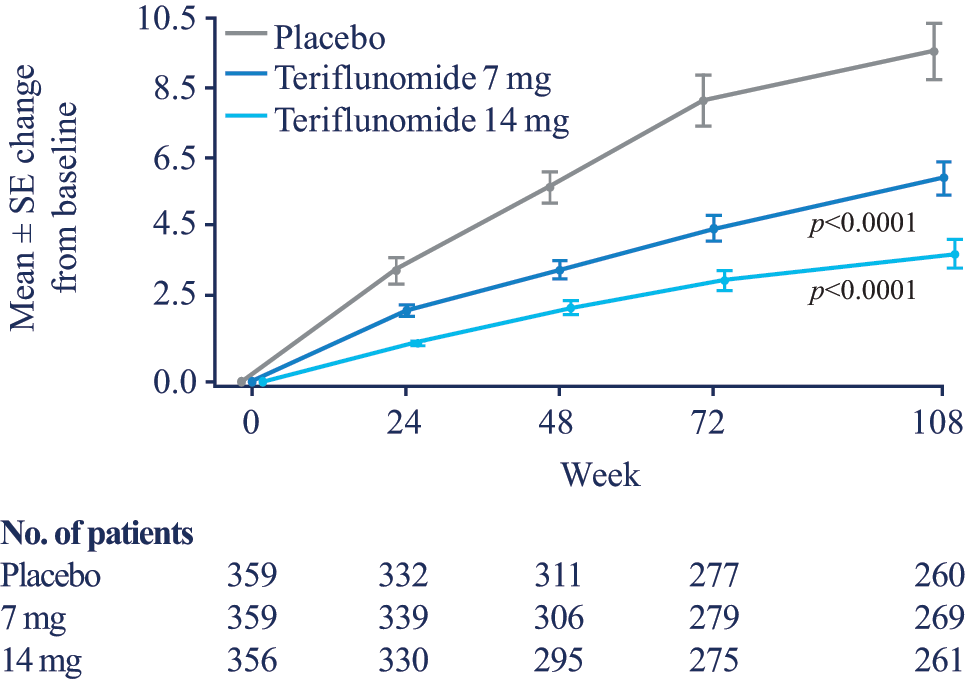
Cumulative number of unique active lesions from baseline.
Z4 score
Significant differences in Z4 score were observed for the 7 mg group (LS mean difference from placebo −0.333 (p=0.0008)) and for the 14 mg group (−0.512 (p<0.0001)) at week 108. The change in Z4 score for both doses statistically diverged from placebo at all times on study (Figure 5). The appearance of the curves and statistical inferences were very similar when Z4 was constructed substituting the Z-transformation of the T2-hyperintense lesion component volume for the total lesion volume in the formula to eliminate any ‘double counting’ (p=0.0007 and p<0.0001 for 7 mg and 14 mg, respectively, compared with placebo).
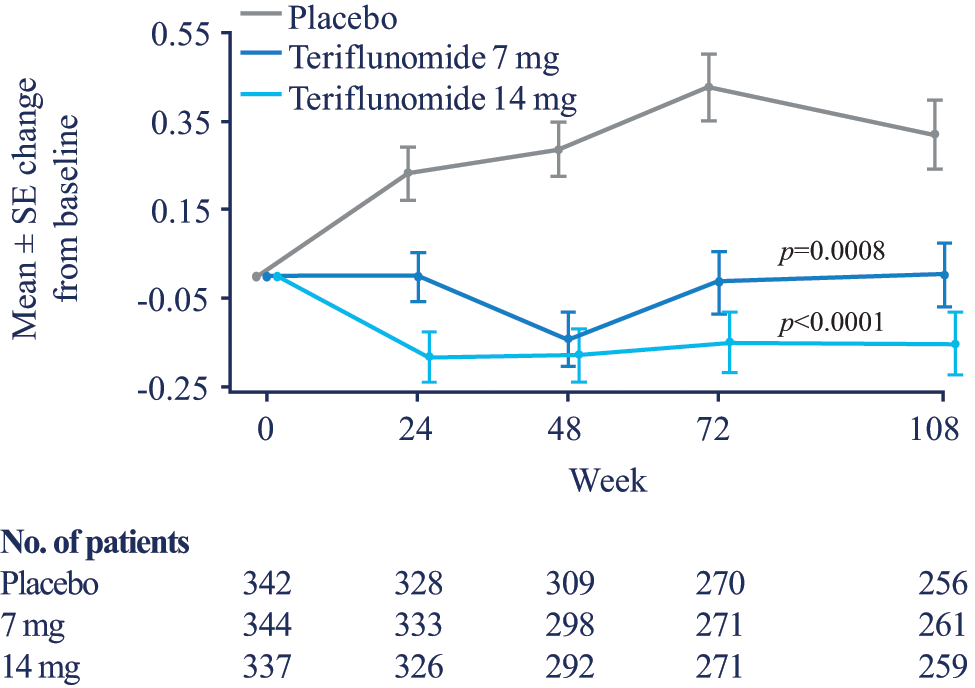
Change from baseline in Z4 score.
Atrophy, volume of GM, volume of WM
Numerically lower BPF reductions occurred over 108 weeks for both teriflunomide groups compared with the placebo group; however, these differences lacked significance. The change from baseline in the volume of GM was also not significant between the treatment groups (LS mean difference from placebo 1.584 for the 7 mg group and −1.985 for the 14 mg group). However, both doses of teriflunomide reduced the loss from baseline in WM volume compared with placebo. The LS mean difference from placebo in WM volume at week 108 was 3.106 (p=0.0609) for 7 mg and 6.146 (p=0.0002) for 14 mg teriflunomide. The actual mean (SD) change in total WM volume from baseline was −4.07±18.34, −1.19±20.56 and 1.23±18.77, for the placebo, 7 mg and 14 mg groups, respectively. This represents a relative change from placebo of 83.0% in the 7 mg and 164.3% in the 14 mg groups (Figure 6).
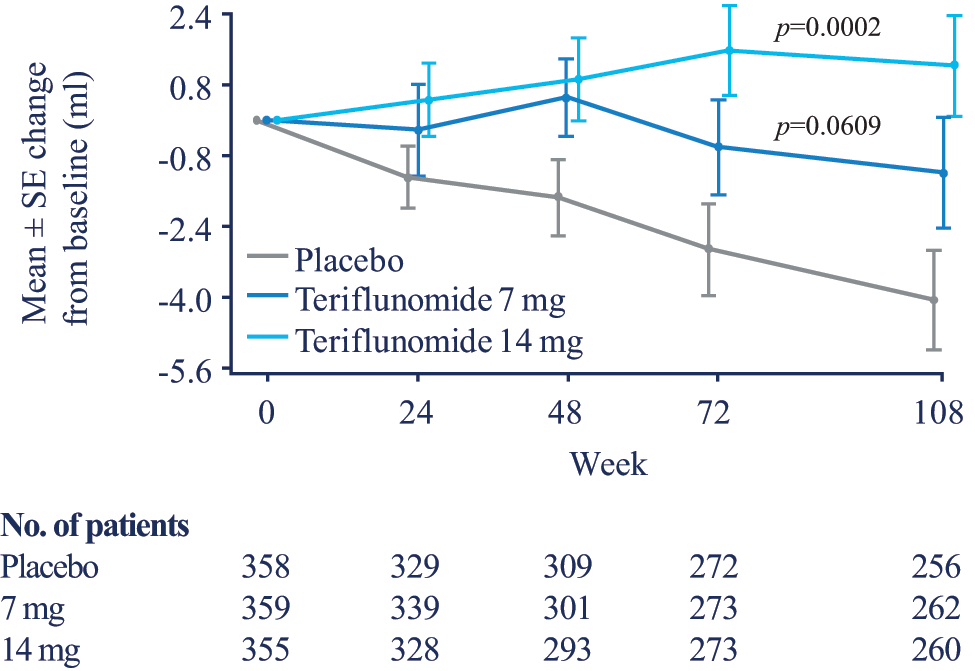
Change from baseline in volume of white matter.
Subgroup analyses
The effects of teriflunomide on total lesion volume, numbers of enhanced lesions and UALs per scan across a number of pre-specified patient subgroups are shown in Figure 7. The effect of teriflunomide relative to placebo was generally consistent on all three MRI measures across all subgroups. Relatively small sample sizes for some subgroups likely contributed to high confidence limits where differences relative to placebo lacked significance.

Effect of teriflunomide on selected magnetic resonance imaging (MRI) outcomes across subgroups.
Discussion
The disease severity of this patient cohort relative to other contemporaneously studied trial cohorts at baseline is worth considering. Literature comparisons can be misleading if the image analysis is performed by different centers using different image-analysis tools. 16 The MRI Analysis Center used the same imaging protocol and segmentation approaches for several relevant trials that can help place the TEMSO cohort in perspective. TOPIC (ClinicalTrials.gov: NCT00622700) is an actively enrolling study of patients randomized within 60–90 days of first MS symptom onset with at least two typical cerebral lesions. The first 430 enrolled patients (mean age 32.1 years; female 66%; monofocal clinical presentation 62%) had a mean total lesion volume of 7.6±8.7 ml, with a T1-hypointense lesion component of 1.4±1.9 ml. The recently completed CombiRx trial (ClinicalTrials.gov: NCT00211887) enrolled 1008 patients with definite relapsing–remitting MS (mean age 37.7 years; female 72%; mean years from diagnosis 1.2±3.3) with a mean total lesion volume of 12.2±13.2 ml, and a T1-hypointense lesion component of 1.7±2.2 ml. 17 This represents a 60% increase in lesion load from the symptom onset to the early diagnosis cohort, and another 60% increase from the early diagnosis cohort to TEMSO, indicating that the TEMSO cohort had more severe MRI-defined cerebral pathology at randomization.
A pre-specified, modified intention-to-treat analysis plan was applied, with the MRI data grouped by original treatment group assignment, and all evaluable data for the MRI measures reported here were used in the analysis as long as the subject remained in the trial. In order to address the issue of the effect that early study dropout might have on the MRI results, a ‘completer’ sensitivity analysis was done that was restricted to only those subjects who completed the 24-month trial. All results were similar (data not shown). The results of this sensitivity analysis make it likely that the effects of teriflunomide on the accumulation of MRI-defined brain pathology are representative and not overly perturbed by the 26.7% overall early clinical termination rate of the clinical study. 7 The effects of both doses of teriflunomide on MRI-defined disease activity (enhanced lesions, UALs and changes in lesion volumes) compared with placebo were readily evident within six months of treatment initiation. The effects of 14 mg teriflunomide were superior to those of 7 mg at most time points. Moreover, the effects of active therapy did not significantly differ based on any of the baseline clinical or MRI characteristics explored in the post-hoc analyses. This suggests that the effects of teriflunomide in attenuating MRI-defined pathologic brain activity are rather independent of disease severity when beginning treatment. The effect of teriflunomide on UALs described here is consistent with the magnitude of the treatment effect observed for on-trial confirmed relapse. A meta-analysis of the effects of interferon beta-1b activity based on new T2 lesions found quite similar relationships between treatment effects on MRI activity measures and relapses. 18 The proportionate effects of teriflunomide on MRI activity and both relapses and trial evidence of accumulating disability also adhere to those described for other DMTs, including other oral therapies. 19 One might anticipate that the dose-dependent effect of teriflunomide on most MRI activity measures would also be reflected in a differential effect on relapse activity. Subsequent analyses of the severity and consequences of relapse observed in this trial support the higher dose as more beneficial (O’Connor et al, submitted) and a clear dose-effect on relapses was also seen in the recently completed TOWER trial (ClinicalTrials.gov: NCT00751881). 20
MRI measures thought to be better indicators of long-term progression (i.e. global atrophy and reduction in GM volume) were not attenuated by teriflunomide. Although teriflunomide reduced the accumulation of the T1-hypointense lesion component volume, this may have been a consequence of the effect of the drug on new activity. That is, the frequency of imaging in this trial favors capturing more new non-enhanced lesion activity than enhanced lesion activity given the rather short-lived duration of most enhancements. Potentially more sensitive measures of atrophy should be explored to determine if any treatment-related effects were missed. Higher resolution, three-dimensional T1-weighted images were acquired from all patients in TEMSO at trial entry, in anticipation that they might prove valuable for additional analysis. Initial samplings suggest that a reasonable proportion of these scans are of adequate quality for delineation of global and regional atrophy, 21 and analyses of cortical thickness, 22 which could further our understanding of the treatment effects in this trial cohort.
An unanticipated and novel finding was the robust effect of teriflunomide in attenuating normal appearing WM loss. This, in part, may relate to the reduction in new lesion activity. However, the impact of teriflunomide therapy on WM appeared disproportional to its effect on activity and changes in total lesion volume. These effects were maintained when the lesion volumes were added back to the normal appearing WM volumes for each scan and the analysis repeated. Thus, the change in tissue segmented as WM likely reflects normalization of the signal characteristics in regions of previously altered tissue. Whether the signal changes reflect tissue repair (e.g. possible remyelination), or other microcellular changes that are not necessarily clinically relevant (such as astrogliosis or microglial cell activation) requires further study. Increased recognition of the amount of lesion ‘resolution’ seen over time by serial image subtraction 23 suggests that the application of this technique to this cohort’s image repository might aid a deeper exploration of the observation.
Teriflunomide provided sustained benefits on brain MRI activity across a range of measures, with a dose effect evident on most parameters. These MRI findings complement clinical data from TEMSO showing significant reductions in relapse rate and disease progression. In addition, the beneficial effect of teriflunomide on MRI endpoints was consistent across selected subgroups in the TEMSO study population. These results, and detailed safety data reported elsewhere, 7 add to the body of evidence suggesting that teriflunomide is a promising new oral monotherapy for RMS and a potential first-line treatment option in this patient population.
Footnotes
TEMSO investigators
AUSTRIA: E Maida – Privatpraxis, Wien; E Auff – Medizinische Universität, Wien; F Fazekas – Medizinische Universität, Graz; T Berger – Medizinische Universität Innsbruck, Innsbruck; CANADA: V Bhan – Queen Elizabeth II (QEII) Health Science Centre, Halifax, NS; J-P Bouchard – Hôpital De l’Enfant Jésus, Québec, QC; P Duquette – CRCHUM, Hôpital Notre-Dame, Montréal, QC; M Freedman – Ottawa General Hospital, Ottawa, ON; F Grand’maison – Clinique Neuro Rive-Sud, Greenfield Park, QC; M Kremenchutzky – London Health Science Centre, London, ON; C Bourque, RA Marrie, M Melanson – Health Sciences Centre, Winnipeg, MB; D Patry – Foothills Medical Centre, Calgary, AB; P O’Connor – St Michael’s Hospital, Toronto, ON; J Oger – Vancouver Hospital - UBC Site, Vancouver, BC; M Stefanelli – Health Sciences Centre, St John’s, NL; F Jacques – Clinique Neuro-Outaouais, Gatineaux, QC; CHILE: P Venegas, M Miranda – Hospital Clinico De la Universidad De Chile, Santiago; N Barrientos – Hospital Dipreca, Santiago; E Tenhamm – Hospital Barros Luco Trudeau, Santiago; S Gloger – Psicomedica, Santiago; G Rohde – Investigaciones Neurologicas, Viña Del Mar; CZECH REPUBLIC: J Mares – Fakultni nemocnice Olomouc, Olomouc; DENMARK: J Frederiksen – Glostrup Hospital, Glostrup; E Stenager – Sygehus Lillebælt, Vejle Sygehus, Vejleand Sygehus Sønderjylland Sønderborg, Sønderborg; ESTONIA: S Haldre – SA Tartu Ülikooli Kliinikum, Tartu; K Gross-Paju – AS Lääne-Tallinna Keskhaigla, Tallinn; FINLAND: I Elovaara, M-L Sumelahti, J-P Erälinna – Suomen Terveystalo, Tampere; J-P Erälinna – Suomen Terveystalo Clinical Research Oy, Turku; M Färkkilä – Postitalon Lääkäriasema, Helsinki; H Harno – Postitalon Lääkäriasema, Helsinki; M Reunanen – Oulu University Hospital/Neurology Clinic, Oulu; T Jolma – Porin Lääkäritalo, Pori; FRANCE: C Confavreux – Hôpital Neurologique Pierre Wertheimer, Lyon; W Camu – CHRU Gui De Chauliac, Montpellier; P Clavelou – Hôpital Gabriel Montpied, Clermont-Ferrand; L Magy – CHRU Hôpital Dupuytren, Limoges; M Debouverie – Hôpital Central, Nancy; G Edan – CHRU Hôpital Pontchaillou, Rennes; C Lebrun-Frenay – CHU Hôpital Pasteur, Nice; T Moreau – CHU de Dijon - Hôpital Général, Dijon; J Pelletier – CHU La Timone, Marseille; E Roullet (deceased) and S Alamowitch – Hôpital Tenon, Paris; M Clanet – Hôpital Purpan, Toulouse; P Hautecoeur – Centre Hospitalier St Philibert, Lomme; P Damier – CHU de Nantes - Hôpital Nord Laënnec, Nantes; L Rumbach – Hôpital Jean Minjoz, Besançon; GERMANY: A Chan, S Schimrigk – St Josef Hospital, Bochum; J Haas – Jüdisches Krankenhaus, Berlin; E Lensch – Deutsche Klinik für Diagnostik, Wiesbaden; H Diener, V Limmroth – Universitätsklinik Essen, Essen; D Anders, M Berghoff, P Oschmann – Universitätsklinikum Gießen, Gießen; M Stangel – Medizinische Hochschule Hannover, Hannover; M Berghoff, A Frese, R Kiefer, M Marziniak – Universitätsklinikum Münster, Münster; U Zettl – Medizinische Fakultät der Universität Rostock, Rostock; E Stark – Klinikum Offenbach GmbH, Hessen, Offenbach; K Jendroska – Neurologische Praxis, Berlin; G Reifschneider – Neuro Zentrum Odenwald, Erbach; ITALY: MP Amato – Azienda Ospedaliero-Universitaria Careggi, Firenze; G Comi – Fondazione Centro San Raffaele del Monte Tabor, Milano; V Cosi – Fondazione Istituto Neurologico Casimiro Mondino, Pavia; P Gallo – Azienda Ospedaliera Di Padova, Padova; C Gasperini – Azienda Ospedaliera San Camillo Forlanini, Roma; A Ghezzi – Azienda Ospedaliera Sant’Antonio Abate di Gallarate, Gallarate (Varese); M Trojano – Azienda Ospedaliero Universitaria Policlinico di Bari, Bari; C Pozzilli – Azienda Ospedaliera Universitaria Policlinico Umberto I, Roma; E Montanari – Presidio Ospedaliero di Fidenza, San Secondo, Fidenza (Parma); NETHERLANDS: CP Zwanikken, PJH Jongen – MS Centrum, Nijmegen; E Th L Van Munster – Jeroen Bosch Ziekenhuis, S-Hertogenbosch; RMM Hupperts – Orbis Medisch Centrum, Sittard-Geleen; B Anten – Orbis Medisch Centrum, Sittard-Geleen; EACM Sanders – Amphia Ziekenhuis, Breda; NORWAY: E Celius – Oslo Universitetssykehus HF, Ullevål, Oslo; H Hovdal – St Olavs Hospital HF, Trondhiem; S Bohne Krogseth – Sykehuset Vestfold HF Tønsberg, Tønsberg; POLAND: W Kozubski – Samodzielny Publiczny Szpital Kliniczny Nr 2, Wielkopolskie, Poznan; H Kwiecinski – Samodzielny Publiczny Centralny Szpital Kliniczny Warszawski Uniwersytet Medyczny, Mazowieckie, Warszawa; A Czlonkowska – Instytut Psychiatrii I Neurologii, Mazowieckie, Warszawa; Z Stelmasiak – Samodzielny Publiczny Szpital Kliniczny Nr 4, Lubelskie, Lublin; K Selmaj – Uniwersytecki Szpital Klinczny, Lódzkie, Lodz; T Hasiec – Wojewodzki Szpital Specjalistyczny SPZOZ, Lubelskie, Lublin; W Fryze – Wojewodzki Szpital Specjalistyczny Im M Kopernika, Pomorskie, Gdansk; W Drozdowski – Samodzielny Publiczny Szpital Kliniczny AM, Podlaskie, Bialystok; J Kochanowicz – NZOZ“KENDRON”, Podlaskie, Bialystok; PORTUGAL: L Cunha – Hospitais da universidade de Coimbra, Coimbra; J De Sá – Hospital De Santa Maria, Lisboa; A Harrington Sena – Hospital De Santo António Dos Capuchos, Lisboa; RUSSIAN FEDERATION: M Odinak – Military Medical Academy Na SM Kirov, St Petersburg; A Skoromets – St Petersburg State Medical University Na IP Pavlov, St Petersburg; E Gusev – City Clinical Hospital #1 Na NI Pirogova, Moscow; A Boiko – Hospital AMO ZIL, Moscow; N Lashch – City Clinical Hospital #11, Moscow; I Stolyarov – Institute Of Human Brain, St Petersburg; A Belova – City Hospital #33, Nizhny Novgorod; N Malkova – Siberian District Medical Center, Novosibirsk; B Doronin – City Clinical Hospital #1, Novosibirsk; E Yakupov – Kazan State Medical University, Kazan; SWEDEN: L Brundin – Karolinska Universitetssjukhuset Solna, Stockholm; J Hillert – Karolinska Universitetssjukhuset Huddinge, Stockholm; SWITZERLAND: L Kappos – University Hospital of Basel, Basel; TURKEY: R Karabudak – Hacettepe Universitesi Tip Fakultesi, Sihhiye, Ankara; C Irkec – Gazi Universitesi Tip Fakultesi, Besevler, Ankara; E Idiman – Dokuz Eylul Universitesi Tip Fakultesi, Izmir; O Turan – Uludag Universitesi Tip Fakultesi, Bursa; H Efendi – Kocaeli Universitesi Tip Fakultesi, Izmit; M Gedizlioglu – SB Izmir Egitim Ve Arastirma Hastanesi, Izmir; UKRAINE: N Buchakchyyska – Zaporozhye Medical Academy of Postgraduate Education, Municipal Institution “Zaporozhye Regional Clinical Hospital”, Zaporozhye; A Goloborodko – Odessa Regional Clinical Hospital, Odessa; A Ipatov – Ukrainian State Scientific-Research Institute of Medical and Social Problems of Disability, Dnipropetrovsk; S Kobets – Ivano-Frankivsk National Medical University, Ivano-Frankivsk Regional Clinical Hospital, Ivano-Frankivsk; V Lebedynets – State Medical and Preventive Treatment Institution “Central Clinical Hospital of Ukrzaliznytsi”, Kharkiv; S Moskovko – Vinnytsya National Medical University NAMI Pirogov, Vinnytsya Regional Psychoneurological Hospital n a academician OI Yushchenko, Vinnytsya; Y Sanotskyy – Lviv Regional Clinical Hospital, Lviv; V Smolanka – Uzhgorod National University, Regional Center of Neurosurgery and Neurology of Uzhgorod, Uzhgorod; V Yavorskaya – Kharkiv Medical Academy of Postgraduate Education, Municipal Institution of Health Care “City Clinical Hospital No. 7”, Kharkov; UNITED KINGDOM: D Bates – Royal Victoria Hospital, Newcastle-upon-Tyne, Northumberland; N Evangelou – Queens Medical Centre, Nottingham, Nottinghamshire; C Hawkins – University Hospital Of North Staffordshire, Stoke-on-Trent, Staffordshire; B McLean – Royal Cornwall Hospital, Truro, Cornwall; J O’Riordan – Ninewells Hospital and Medical School, Dundee, Scotland; S Price – Royal Hallamshire Hospital, Sheffield, South Yorkshire; B Turner – the Royal London Hospital, London; D Barnes – St George’s Hospital, Department of Neurology, London; J Zajicek – Memory Assessment Centre, Plymouth, Devon; UNITED STATES: W Honeycutt – Neurology Associates PA, Maitland, FL; O Khan – Wayne State University School of Medicine, Detroit, MI; L Spikol – Lehigh Valley Hospital, Allentown, PA; J Stevens – Fort Wayne Neurological Center, Fort Wayne, IN.
Conflicts of interest
JW has received consulting fees or honoraria from sanofi-aventis for his role as a steering committee member for the clinical trial development program. Travel support received from sanofi-aventis in relation to steering committee membership, consultation activities related to trial program and investigator trial meetings. Board membership (BC Decker, Eli Lilly, Novartis, sanofi-aventis, Teva and UCB) and consultancy (Acorda, Actelion, Astellas, Bayer Health Care, Celgene, Genetech, Genzyme, (a Sanofi company) Hoffman La Roche, Jansen RND, Novartis, Sanofi, Teva and Teva Neuroscience). Payment for lectures including service on speaker bureaus (Biogen Idec, Consortium MS Centers, EMD Serono, Medscape CME, Pfizer, Serono Symposia International Foundation, SUNY Stony Brook Foundation, Teva, Texas Neurological Society, University of Buffalo, University of Utah, USF Health Professionals and UTMB). The institution at which JW works has received royalties from Millipore (Chemicon International) Corporation.
PAN has received consulting fees from Genzyme (a Sanofi company) and Acorda Pharmaceuticals.
FN has been a member of an Advisory Board for Genzyme (a Sanofi company).
SD has no conflicts of interest to report.
POC has received consulting fees and/or research support from Actelion, Bayer, Biogen Idec, BioMS, Cognosci, Daiichi Sankyo, EMD Serono, Genentech, Genmab, Genzyme (a Sanofi company), Novartis, Roche, sanofi-aventis, Teva and Warburg Pincus.
CC has received consulting fees from Biogen Dompe, Biogen Idec, Gemacbio, Genzyme (a Sanofi company), Hertie Foundation, Novartis, sanofi-aventis, Teva and UCB; lecture fees from Bayer-Schering, Biogen Idec, Genzyme (a Sanofi company), Merck-Serono, Novartis, Octopharma, sanofi-aventis and Teva; and research support from Bayer-Schering, Biogen Idec, Merck Serono, Novartis, sanofi-aventis and Teva.
GC has received compensation for consulting services and/or speaking activities from Actelion, Bayer Schering Pharma AG, Biogen, Genzyme (a Sanofi company), Merck Serono International, Novartis, Sanofi, Serono Symposia International Foundation and Teva.
LK has received research support from Actelion, Advancell, Allozyne, Bayer Health Care Pharmaceuticals, Bayer Schering Pharma, Bayhill, Biogen Idec, BioMarin, CLC Behring, Elan, Genmab, Genmark, GeNeuro SA, GlaxoSmithKline, Lilly, Merck Serono, Novartis, Peptimmune, Sanofi, Santhera, Roche, Teva, UCB and Wyeth.
TPO has received lecture and advisory board honoraria from Bayer, Biogen Idec, Genzyme (a Sanofi company), Merck and Novartis. He has also received unrestricted research grants from Bayer, Biogen Idec, Merck, Novartis and sanofi-aventis.
PT works as an employee of the sponsor.
LW works as an employee of the sponsor.
AM has received consulting fees and travel support from sanofi-aventis for his role as a steering committee member for the clinical trial development program of teriflunomide. He has also served as a consultant and/or member of advisory boards for Accordant Health Services, Acorda, Biogen Idec, EMD Serono, Genzyme (a Sanofi company), GlaxoSmithKline, Merck Serono, Novartis, Nuron Biotech, ONO, Questcor, and Teva Neurosciences. He has received research support from Acorda, Biogen Idec, Genentech, Genzyme (a Sanofi company), Novartis, Roche and sanofi-aventis. He is not a member of a Speakers’ Bureau and holds no stock in any relevant companies.
MF has received personal compensation as an advisor/consultant/steering committee member or speaker for Bayer HealthCare, Biogen Idec, Celgene, Merck Serono, Novartis, sanofi-aventis and Teva Pharma; and research support from Bayer and Genzyme (a Sanofi company).
Funding
The study was supported by sanofi-aventis. Editorial assistance was provided by Fishawack Communications Ltd, funded by sanofi-aventis. In collaboration with the TEMSO Trial Group, sanofi-aventis designed the study, analyzed and interpreted the data, and edited the report. Data were recorded at participating clinical centers, and then collected and maintained by sanofi-aventis. All authors had full access to the data. The authors had final responsibility for the decision to submit for publication.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.