
Abstract
Keywords
Introduction
Multiple sclerosis (MS) is an inflammatory autoimmune disease that targets myelin-associated antigens, resulting in neurodegeneration and axonal loss within the central nervous system (CNS). 1 MS symptoms usually begin with relapses and remissions (RR-MS), and neurologic deficits accumulate over time with secondary progression (SP-MS). 2 A number of immunomodulatory and immunosuppressive drugs have been approved for the treatment of MS. 3 –5 However, none of these disease-modifying drugs is a ‘cure’, and each displays the potential for serious adverse events. Moreover, issues of tolerability limit treatment for many patients. 6 Thus, an effective therapy with an improved safety and tolerability profile remains an important unmet need for patients afflicted with multiple sclerosis.
Myelin reactive T-cells (MRTCs) have been strongly implicated in the neuropathogenesis of MS. 7,8 Autologous T-cell immunotherapy uses an ex vivo enriched source of human MRTC, attenuated by irradiation, as the treatment agent to sensitize the immune system in subjects with MS to deplete and/or regulate pathogenic myelin reactive T-cells that maintain autoimmune processes within the CNS. 9 –13 Tovaxin® is an autologous T-cell immunotherapy that consists of a pool of up to six T-cell lines raised against immunodominant peptides derived from myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG) and proteolipid protein (PLP). The immunodominant peptides are selected on a per subject basis by pre-screening for reactivity against overlapping peptide libraries covering the three major myelin antigens. Utilizing this approach, Tovaxin® is a personalized product matched to the anti-myelin T-cell repertoires of individual subjects with MS.
Early phase clinical studies with Tovaxin® were conducted in subjects with RR-MS and SP-MS who were intolerant of, or unresponsive to, other MS treatments. Results indicated that Tovaxin® was safe and well tolerated, and appeared to be associated with clinical benefit. In subjects with active disease, as indicated by a mean annualized relapse rate (ARR) of 1.3 at study entry, a 64–85% reduction in ARR was seen at 1 year. 14 In the light of encouraging data from the uncontrolled Phase 1/2 program, a Phase 2b randomized double-blind placebo-controlled study (Tovaxin for Early Relapsing Multiple Sclerosis, TERMS) was conducted to further evaluate safety, tolerability and efficacy of Tovaxin® in subjects with RR-MS or clinically isolated syndrome (CIS). Of the 150 subjects enrolled, a subset with RR-MS with more severe clinical disease, defined a priori as ARR > 1 at study entry, was identified prospectively for analyses (n = 50). Further post-hoc analysis was performed on subjects naïve to disease-modifying treatments (DMT) prior to study entry. The conclusions from these analyses highlight the importance of studying a population of subjects with more active MS, particularly in the absence of prior disease-modifying treatments, to detect meaningful clinical differences between groups of limited size, and in studies of short duration.
Methods
Ethics statement
This study was conducted in accordance with the Declaration of Helsinki, and approval by a centralized ethic review committee or academic institutional review board (IRB) was obtained where participants were recruited and human experimentation was conducted in this multi-center clinical trial (NCT00245622). All patients provided written informed consent prior to study entry. This clinical trial was designed and reported according to recommendations of the Consolidated Standard of Reporting Trials (CONSORT) statement.
Preparation of Tovaxin®
MS subjects were screened for the presence of detectable MRTC in the peripheral blood using a T-cell proliferation assay. In brief, 2 × 105 peripheral blood mononuclear cells (PBMC) were set up in triplicate cultures on day 0 with pools of two peptides from peptide libraries spanning MBP, MOG, and PLP and cultured in the presence of 20 U/ml interleukin (IL)-2. Individual peptides were 16-mers with the library generated using a 12-mer overlap. In total, 54 peptide pools were evaluated, representing the three myelin antigens. On day 6, cultures were pulsed with 0.5 μCi of tritiated thymidine, and proliferation determined after a further 18 h in culture. Positive proliferation to any peptide pool was defined by a stimulation index of ≥2 versus control cells cultured in media alone. Once up to six reactive peptide pools had been selected, a unit of whole blood was procured in accordance with the IRB-approved signed informed consent prior to study screening. Autologous MRTC were expanded from PBMC with four successive weekly peptide (20 μg/ml) re-stimulations in the presence of autologous antigen presenting cells and IL-2 (20 U/ml). MRTC cell lines were then individually cryopreserved for the future production of final doses of Tovaxin® to meet clinical dosing requirements. Fourteen days prior to dosing, autologous cryopreserved cell lines were thawed and expanded in the presence of 5 μg/ml phytohemagglutinin (PHA) and IL-2 (100 U/ml), and pooled to achieve a target dose of 30–45 × 106 viable cells. Each dose of Tovaxin® was formulated in 2 ml 4% human serum albumin (HSA) in sterile saline, then attenuated by irradiation (10,000 Rads) and subjected to sterility, endotoxin and mycoplasma testing prior to final product release. All products were required to meet a predefined release specification of ≥85% CD3+ T-cells. The mean T-cell content across all Tovaxin® products manufactured was 95%. The CD4+ and CD8+ T-cell content in Tovaxin® can vary between MS subjects, depending on the degree of HLA class I and II restricted immunity to peptides when up to six cell lines are pooled to generate the final doses. However, repetitive expansion of cell lines over time from any one subject’s cell lines generated comparable Tovaxin® final products in terms of the ratio of CD4+ to CD8+ T-cells per dose.
Preparation of placebo
Placebo was formulated with only inactive components (HSA and sterile saline) and packaged in the same way as Tovaxin® to protect the study blind. All manufacturing processes were conducted under appropriate cGMP/GTP regulations.
Protocol design
This study was designed to enroll a total of approximately 150 subjects, males and females between 18 and 55 years of age inclusive at screening, at 30–40 different sites in the United States.
Eligible candidates were randomized to receive either Tovaxin® or placebo, in a 2:1 ratio. Inclusion criteria included the presence of MRTC at screening; and either CIS suggestive of CNS demyelination within 12 months of screening, or a diagnosis of MS within the past 10 years with a clinical course consistent with RR-MS accompanied by a specific MRI result at screening (one Gd+ lesion, or two lesions consistent with MS), or one relapse within 12 months or two within 24 months; and screening and baseline EDSS scores between 0.0 and 5.5, inclusive. Exclusion criteria included a history of cancer, HIV, or other serious illness; ongoing disease-modifying treatment for MS during the 30 days prior to screening; treatment with systemic corticosteroids within 60 days prior to screening; diagnosis of progressive-stage MS; prior treatment with total lymphoid irradiation or cladribine; and T-cell or T-cell receptor immunotherapy. Tovaxin® or placebo was administered as five subcutaneous injections (five visits, with one dose divided into two injections; 1 ml in each arm at each visit) by unblinded study personnel at weeks 0, 4, 8, 12, and 24. After each treatment, the subject remained in the clinic for 1 h, and was later contacted by phone the following day, to monitor for latent injection related adverse effects.
Randomization and blinding
All 150 subjects enrolled were screened according to the clinical protocol. Eligible subjects were stratified by MRI gadolinium lesion status (Gd+ or Gd−) and disease course, and then randomized to either the Tovaxin® or placebo arm of the study in blocks of six by an unblinded representative.
A designated blinded investigator and study coordinator performed neurological examinations and gathered study data. The subject was instructed to refrain from discussing any injection site reactions with the blinded PI. An unblinded physician evaluated and treated injection site reactions.
Sample size
Using data from previous studies, it was assumed the subjects receiving placebo would have a mean (± standard deviation [SD]) number of contrast-enhancing lesions of 8.3 ± 12.37 after four scans, and that Tovaxin® treatment would result in a reduction in that number of approximately 50% over weeks 28, 36, 44, and 52. Using 80% power, a two-sided significance level of 0.05, 2:1 randomization, and with baseline correction, a minimum number of 90 evaluable subjects in the Tovaxin® arm and 45 in the placebo arm were required. Assuming a dropout rate of 10%, the actual number of enrolled subjects necessary was determined to be approximately 100 subjects in the Tovaxin® arm and 50 in the placebo arm.
Analysis populations
The modified Intent to Treat population (mITT) consisted of all subjects who received at least one dose of study product and had at least one MRI scan at week 28 or later.
Of the 150 enrolled subjects, 142 comprised the population for the analysis of the primary and secondary endpoints. An analysis of 50 out of 150 subjects with more clinically active MS, defined as baseline ARR > 1, which included those subjects who had two or more relapses within 12 months prior to screening, was specified in the analysis plan for the study prior to study unblinding. Baseline disease characteristics for this subgroup compared with the study population as a whole were similar (Table 1).
Baseline Disease Characteristics by Study Population.
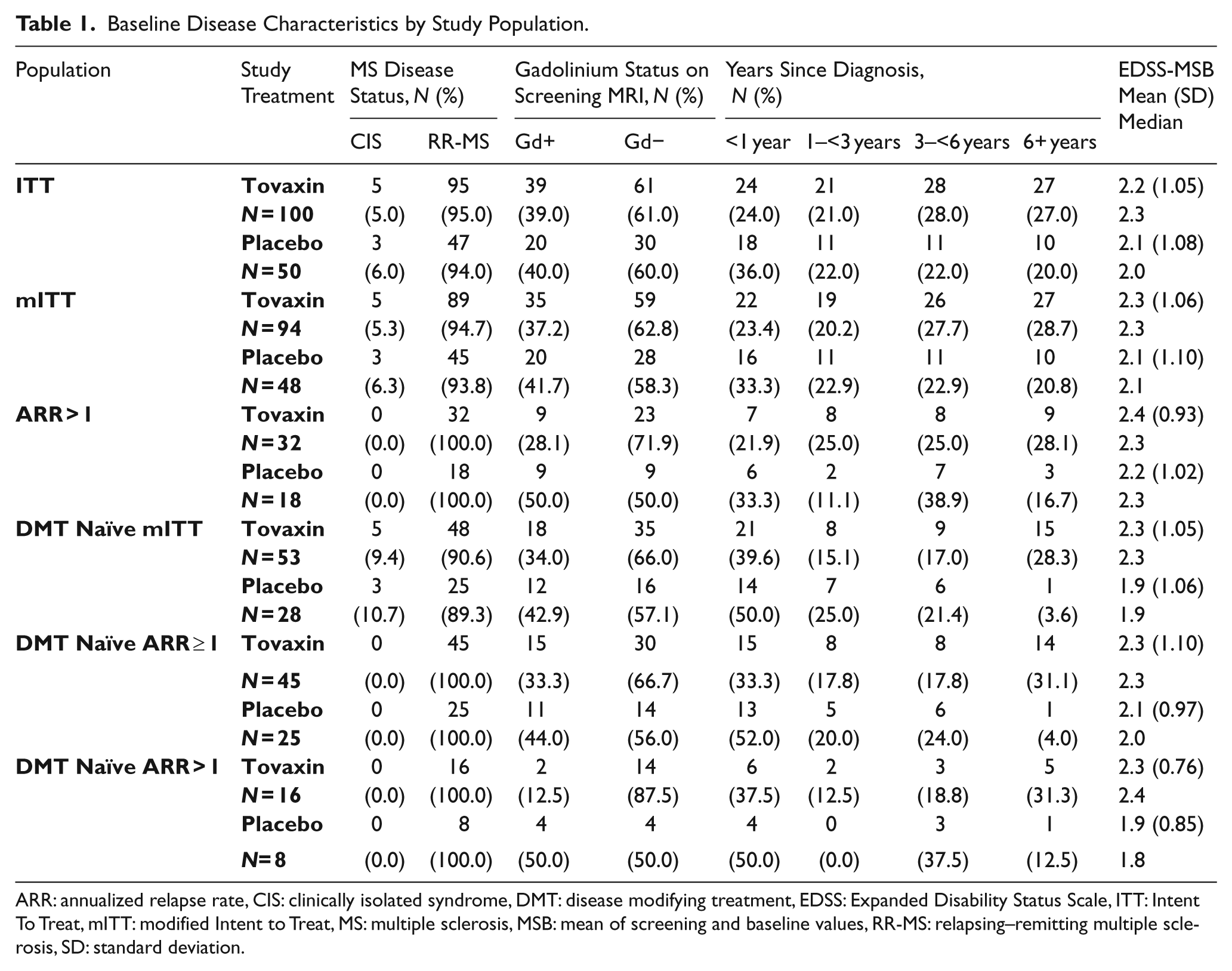
ARR: annualized relapse rate, CIS: clinically isolated syndrome, DMT: disease modifying treatment, EDSS: Expanded Disability Status Scale, ITT: Intent To Treat, mITT: modified Intent to Treat, MS: multiple sclerosis, MSB: mean of screening and baseline values, RR-MS: relapsing–remitting multiple sclerosis, SD: standard deviation.
Given the Phase 2 nature of this study, post-hoc analysis was performed to expose cofactors that may have contributed to the study results. These exploratory analyses were restricted to the prognostic factors of baseline ARR, EDSS, and Gd+ status as well as previous DMT use.
Post-hoc analysis was performed on subjects naïve to disease-modifying treatments prior to joining the TERMS study. Three subgroups were chosen: the mITT, the prospective ARR > 1 and a third subgroup consisting of subjects defined as ARR ≥ 1, which included those subjects who either met the ARR > 1 relapse criteria or had had just one relapse in the 12 months prior to screening. The naïve mITT population was comprised of 81 subjects, the ARR > 1 was comprised of 24 subjects and the ARR ≥ 1 was comprised of 70 subjects.
Study endpoints
The primary endpoint of this study was the cumulative number of gadolinium-enhancing lesions on T1-weighted MRI summed over weeks 28, 36, 44, and 52.
The secondary endpoints were cumulative number of new gadolinium-enhancing lesions at weeks 28–52, change in T2-weighted lesion volume and ARR at the end of the 1-year study. In addition, disability was monitored according to the Expanded Disability Status Scale (EDSS).
Radiographical measures
MRIs were completed at screening, baseline, during the treatment phase (weeks 8 and 12) and during the efficacy phase (weeks 28, 36, 44, and 52) using a standard MRI protocol and centralized reading center. Brain atrophy was measured by calculating change in brain volume from baseline to the last MRI performed in the study (week 28 through week 52) using the SIENA method.
Protocol defined relapse
For the purposes of this trial, a relapse was defined as an onset of new or recurring neurological signs and/or symptoms associated with MS, or worsening of pre-existing symptoms associated with MS, in the absence of febrile illness or steroid withdrawal reactions. The signs and/or symptoms must have: persisted for 24 h or more; followed a period of symptomatic stability or improvement that was at least 30 days in duration; been accompanied by new, objective neurological findings observed by a blinded investigator; and been associated with a documented increase in the EDSS of at least 1.0 point at the time of relapse evaluation. Data from protocol defined and investigator defined relapses were both captured during the study.
Disability assessment
The EDSS score was utilized with a complete neurological examination at screening, baseline, and eight additional time points. The EDSS was performed at each site by the same blinded individual. The entry EDSS reported is the mean of the screening and baseline (MSB) values.
Safety assessments
Adverse events (AE), serious AEs (SAEs), treatment discontinuations due to AEs, injection site AEs, complete blood count with differentials, serum blood chemistries, urinalysis, vital signs, physical exams, and neurological exams were evaluated.
Statistical methods
Radiographic outcomes included the cumulative number of enhancing lesions on T1-weighted MRIs, summed over the week 28, 36, 44, and 52 MRIs, the number of new enhancing lesions at weeks 28–52, and the change from baseline in T2-weighted lesion volume at week 52 between the Tovaxin® and placebo groups. The planned analyses for these were adjusted for the number of enhancing lesions at baseline. The adjustment was established by calculating the mean of the number of enhancing lesions on T1-weighted MRIs at screening (visit 1) and baseline (visit 3) MRIs (MSB). Analyses were also stratified by presence or absence of at least one enhancing lesion (Gd+/−) at screening. A nonparametric analysis of covariance (ANCOVA), van Elteren test incorporating stratification based on Gd+/− status on the screening MRI and covariate adjustment based on the ranked number of enhancing lesions at baseline (MSB) were used to compare the treatment arm’s responses.
The group ARR was calculated as: (Total number of relapses in the period for all subjects/Total number of subject days in the period) × 365.25. The analysis included relapses meeting protocol defined criteria for a relapse. The Wald test with stratification by baseline Gd+/− status and baseline ARR (≤1, >1) and an overdispersed Poisson regression were noted in the analysis plan as analysis methods for the annualized relapse rates. A subsequent publication recommended negative binomial regression as the best technique for the analysis of the overdispersed Poisson distributed variable, relapse rate. 15 Therefore, the negative binomial method, without adjustment for strata, is presented for all such analyses in this paper. The treatment effect and a dispersion parameter were used to model the number of relapses with an offset variable reflecting the log of the duration over which the subject was followed.
Changes in clinical disability, including worsening, no change, and improvement (beneficial), defined by at least 1 point EDSS score change from baseline, are reported. The analysis of the absolute change in EDSS scores from baseline to week 52 was a non parametric ANCOVA, as was performed for the radiological outcomes, was a nonparametric ANCOVA. The Cochran–Mantel–Haenszel test was used to evaluate the equality of distribution of EDSS categories at the end of the study stratified by Gd+/− on screening MRI and on the number of contrast enhancing lesions (CEL) CEL (0, 1–5, >5) at baseline (MSB). Unless otherwise stated, all statistical tests were two-sided with the probability of a Type 1 error not to exceed α = 0.05 for the null hypothesis of no difference between the treatment groups. The incidence of adverse events was analyzed descriptively.
The ITT population was used for safety analyses and consisted of all subjects who received at least one dose of the randomized study treatment as assigned. The mITT population was used for primary and secondary endpoint analyses and consisted of all subjects in the ITT set who had at least one MRI scan at week 28 or later (weeks 36, 44, or 52).
Results
Study populations
A total of 304 subjects were screened, 152 were randomized, and 150 received study treatment: 100 were treated with Tovaxin® and 50 with placebo at 33 US centers between 2 November 2006 and 30 August 2007. The mean age of study participants was 39.0 years, with 36 males and 114 females. Age and gender were comparably distributed between active and control in all study populations (Supplemental Table 1). The flow of subjects from screening to study completion and analysis is presented in Figure 1, and radiologic and clinical status for all analysis sets at study entry is displayed in Table 1.
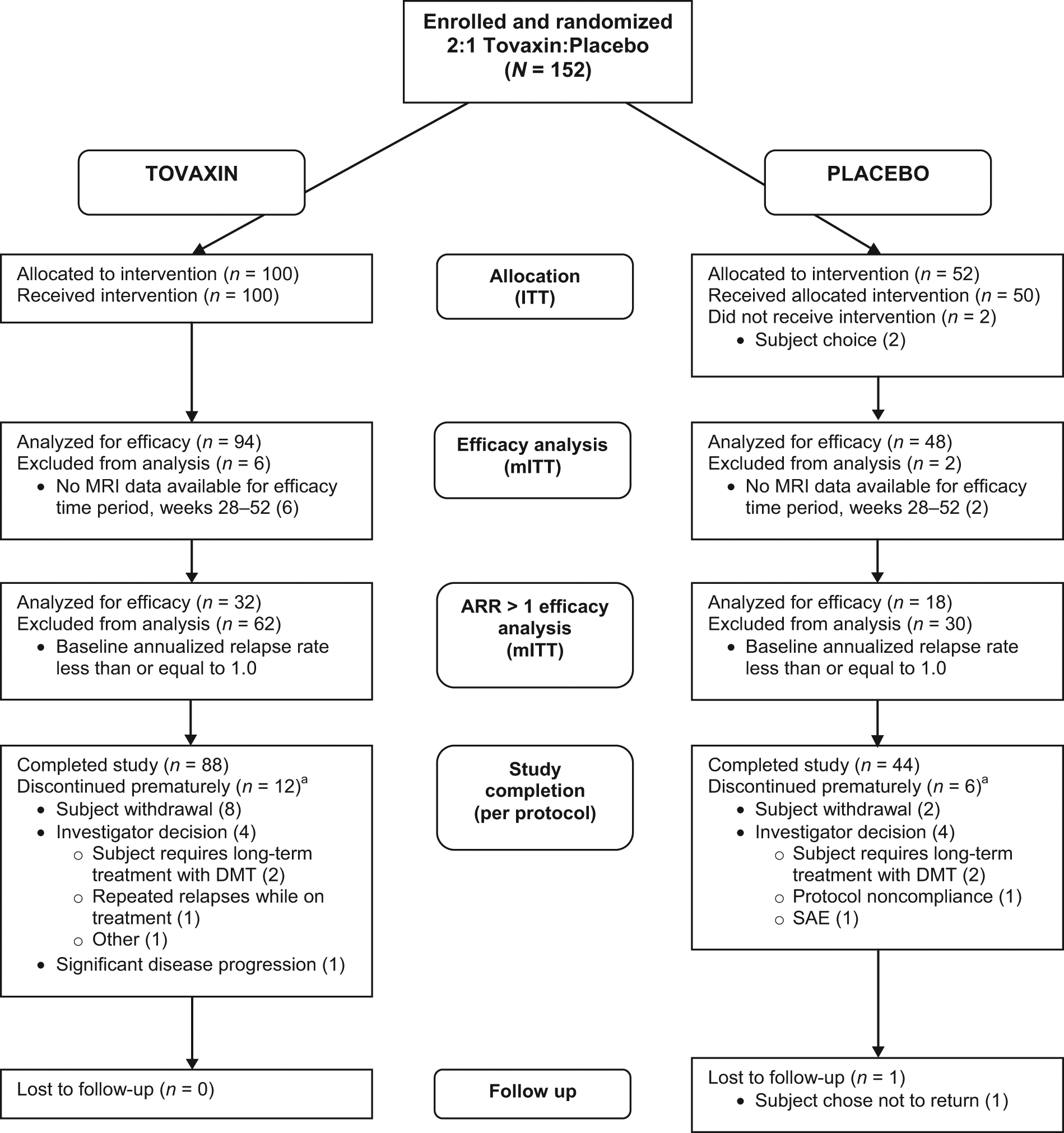
CONSORT flow diagram.
The 150 subjects who received at least one study treatment, representing the ITT population, were monitored for safety throughout the study and used for the safety analysis. The mITT set consists of all subjects in the ITT who had at least one MRI during the endpoint time period (week 28 through week 52). These were analyzed in the treatment group to which they were randomized: 94 were treated with Tovaxin® and 48 with placebo. Previous DMT exposure was equally distributed between Tovaxin® and placebo treatment groups: 44% vs. 42%, respectively. The mITT set was used for the analysis of primary and secondary endpoints. A prospective subset of the total mITT included those with more active disease, with an ARR > 1, and was populated according to individual subject baseline ARR values. Among these subjects, 32 were treated with Tovaxin® and 18 with placebo. The distribution of DMT exposure in the ARR > 1 subset was similar to that in the larger mITT population, with 50% Tovaxin® and 56% placebo subjects having prior exposure. The mITT, ARR > 1 and ARR ≥ 1 groups were also included in the smaller treatment naïve subgroups used for post-hoc analysis of clinical efficacy, and were populated according to the absence of any previous exposure to DMT for MS prior to study enrollment. Prior DMT exposure in the non-naïve group included: azathioprine, methotrexate, glatiramer acetate, interferon beta, intravenous immunoglobulin, natalizumab, alemtuzumab, daclizimab, rituximab, naltrexone or mitoxantrone (Supplemental Table 2). The mITT DMT naïve subset (n = 81) consisted of 53 treated with Tovaxin® and 28 with placebo. The ARR > 1 subset (n = 24) consisted of 16 treated with Tovaxin® and eight with placebo. The ARR ≥ 1 subset (n = 70) consisted of 45 treated with Tovaxin® and 25 with placebo.
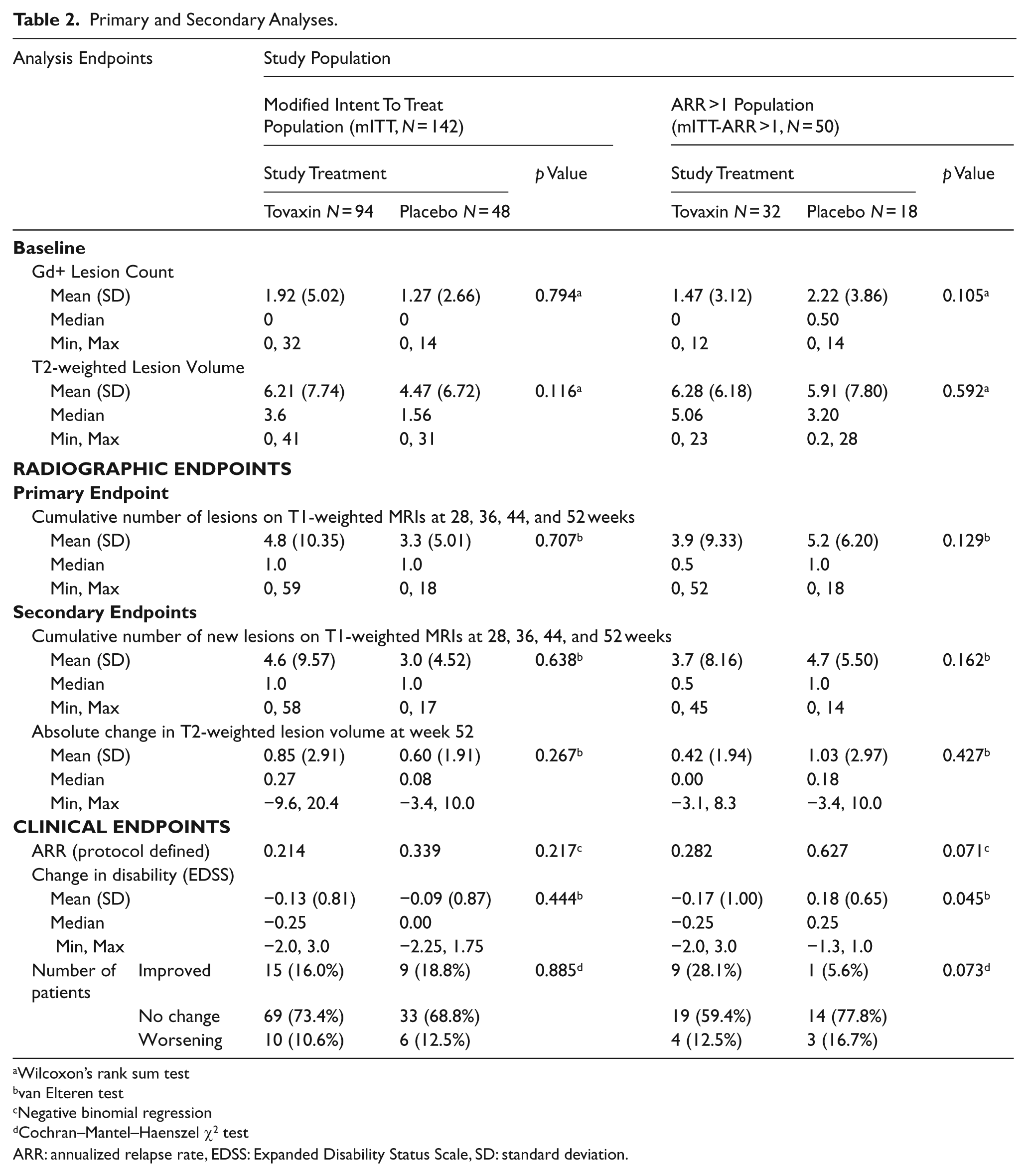
Primary and Secondary Analyses.
Wilcoxon’s rank sum test
van Elteren test
Negative binomial regression
Cochran–Mantel–Haenszel χ2 test
ARR: annualized relapse rate, EDSS: Expanded Disability Status Scale, SD: standard deviation.
Safety analysis
All 150 subjects in the ITT population were monitored for safety. Safety results for each subgroup were essentially identical to those for the ITT population as a whole. There were no deaths on the study, no discontinuations due to AEs, and no SAEs that, in the opinion of the investigators, were related to Tovaxin® treatment. Most of the reported AEs in this study involved local reaction to study treatment injection. Of the 100 subjects who received Tovaxin®, 84 reported 363 injection site events, and 33 of the 50 subjects who received placebo reported 122 events. The most commonly reported AEs in both groups were: erythema (56 [56%] subjects receiving Tovaxin® vs. 19 [38%] placebo), bruising (41 [(41%] receiving Tovaxin® vs. 16 [32%] placebo), pain (36 [36%] in the Tovaxin® group vs. 12 [24%] placebo), and swelling (29 [29%] in the Tovaxin® group vs. 14 [28%] placebo). Of the 100 subjects receiving Tovaxin®, 63 reported injection reactions that were definitely, probably, or possibly related to study treatment, with erythema being the most common (38 subjects). Of the 50 placebo subjects, 26 subjects reported events which may have been related to study treatment, with erythema the most frequently reported related AE (18 subjects).
Five subjects treated with Tovaxin® reported a total of five SAEs (five events), and nine subjects treated with placebo reported 13 SAEs. These events were listed as SAEs due to the need of inpatient treatment, but they were not severe in intensity or considered by principal investigators or study physicians to be related to study treatment. In the ITT population as a whole, SAEs among Tovaxin®-treated subjects included diplopia, lower limb fracture, arthralgia, intervertebral disc degeneration, and muscular weakness. Among placebo-treated subjects, the 13 SAEs included gastroesophageal reflux disease, Mallory–Weiss syndrome, non-cardiac chest pain, gastroenteritis, Helicobacter gastritis, pneumonia, urinary tract infection, extremity pain, convulsion, myoclonic epilepsy, nystagmus, a suicide attempt, and menorrhagia.
Modified Intent to Treat analysis
In the mITT population, consisting of 142 treated subjects, no statistically significant differences between subjects administered Tovaxin® and those administered placebo were observed for clinical or radiographic endpoints (Table 2).
Prospective ARR > 1 subgroup analysis
The TERMS study was conducted over a period of 12 months. The ability to detect clinical benefit over a relatively short time frame may be enhanced by studying those subjects with more active disease, in particular with annualized relapse histories of more than one relapse per year. Therefore, a prospective efficacy analysis of subjects with ARR > 1 at baseline (n = 50) was included in the statistical analysis plan (Table 2). The group ARR for the Tovaxin®-treated subjects trended lower than those subjects treated with placebo (0.28 for Tovaxin® vs. 0.63 for placebo, p = 0.071), resulting in a 56% reduction in relapses in the Tovaxin®-treated subjects compared with placebo. In addition, improvements were noted in the key measure of disability: the change in mean EDSS scores (−0.17 ± 1.00 for Tovaxin® vs. 0.18 ± 0.65 for placebo, p = 0.045). An overall improvement in disability was experienced by 28.1% of the Tovaxin®-treated subjects compared with 5.6% of placebo subjects (p = 0.073). Despite evidence of positive clinical indicators in favor of Tovaxin® as defined by ARR and EDSS, no clear improvements in radiologic endpoints were observed.
Treatment naïve subgroup analysis
Various prognostic factors were investigated to determine their influence on the key outcomes. Although the TERMS protocol allowed for a washout period of at least 30 days for prior disease-modifying agents and 60 days for systemic corticosteroids for the treatment of relapse before entering the study, post-hoc analysis was conducted on DMT naïve subjects to explore the possible impact of prior therapies on ARR, EDSS, and MRI outcomes in three sub-populations. An analysis was performed on subjects in the mITT and the prospective ARR > 1 groups and those with an ARR ≥ 1 at baseline (Figure 2). In the mITT population, the Tovaxin®-treated subjects had an ARR of 0.21, 56% lower than the placebo ARR of 0.48 (p = 0.060). On completion of the study, the Tovaxin®-treated subjects in the ARR ≥ 1 population had an ARR of 0.18, 64% lower than the placebo ARR of 0.50 (p = 0.046). In the most active disease group (ARR > 1), the Tovaxin®-treated subjects had an ARR of 0.31, 73% lower than the placebo ARR of 1.18 (p = 0.009). Although Tovaxin®-dependent improvements in ARR were recorded, particularly in those subjects with more active disease, no clear improvements were noted in either EDSS or radiologic measures in these relatively small subgroups.
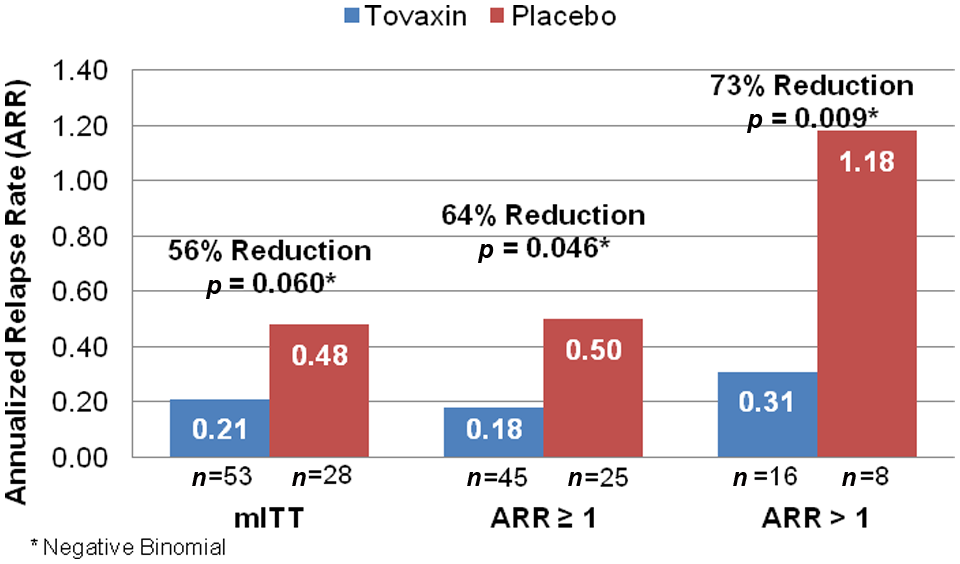
Annualized relapse rate in DMT naïve populations.
Discussion
Tovaxin® was shown to be reasonably safe and well tolerated when administered over the course of 24 weeks in subjects with CIS and in those with RR-MS, including subjects with more active clinical disease at study entry. No deaths occurred, and no reported SAEs, in the opinion of the investigators, were associated with treatment. The most common adverse events were mild injection site reactions.
Results of clinical efficacy and radiographic analyses among the 142-subject mITT population demonstrated no statistically significant differences in outcomes between Tovaxin® and placebo groups. The mITT contained subjects with diverse clinical histories, from CIS to RR-MS, including those with more active disease, namely ARR > 1. Both radiologic and clinical data were accumulated over a 12-month period from baseline. The clinically diverse mITT population and a low placebo ARR of 0.34 observed in TERMS resulted in the study being underpowered to detect positive clinical benefit within the mITT population. This outcome was also likely influenced by the relatively short time frame of the trial, namely 12 months of observation from baseline.
Data from the previous Phase 1/2 trial indicated that Tovaxin® showed a clinical benefit in subjects with more severe disease, as defined by mean ARR > 1 at study entry. 14 Therefore, a prospective analysis of subjects with ARR > 1 was included in the TERMS statistical analysis plan. In this prospective subgroup analysis, clinical assessments favored Tovaxin® in this population of subjects with more active RR-MS. A positive trend, as defined by a reduction in ARR for the Tovaxin®-treated group versus placebo (p = 0.071) and stabilization or improvement in EDSS from baseline (p = 0.045), was demonstrated.
An ARR of 0.34 in the placebo mITT population stimulated further post-hoc analysis of the data set to identify important prognostic factors that may be associated with meaningful differences between Tovaxin® and placebo treated subjects. Subjects naïve to previous DMT were selected on the hypothesis that exposure to alternate therapies may have imparted a protective ‘legacy effect’, reducing disease activity despite the protocol defined washout period of 30 days prior to study entry for DMT and 60 days for systemic corticosteroid treatment for relapse. Among subjects naïve to DMT, the ARR in the placebo group was 0.48, considerably larger than the ARR found in the placebo mITT population (0.34). Analysis of subject demographics, clinical and radiological parameters at study entry showed that these factors could not account for the observed differences in ARR for placebo subjects on the TERMS protocol, when subgrouped on the basis of the presence or absence of prior DMT therapy.
To formally examine the impact of prior DMT on the TERMS data set, post-hoc analysis was conducted on the mITT group and extended to two additional sets, namely ARR > 1 and a larger subset with ARR ≥ 1, all of whom were naïve to DMT before joining the TERMS study. The data showed a positive trend in favor of Tovaxin® as defined by ARR for the mITT group as a whole, and in both subgroups, ARR > 1 and ARR ≥ 1 (Figure 2). However, these results should be interpreted with caution due to the post-hoc nature of these analyses.
The impact of prior therapies on the placebo group would indicate that, despite a washout period to remove potential DMTs as defined by pharmacokinetics, a ‘legacy effect’ might persist. Cytotoxic drugs, depleting antibodies or agents designed to influence the homing of leukocytes have been shown to actively disable the immune response for prolonged periods beyond the pharmacokinetic washout period of the drug before immune reconstitution is achieved. 16 –18 In addition, immunomodulators such as interferon beta or glatiramer acetate have been shown to influence the immune response to myelin antigen through several active processes, which may persist in the absence of therapy. 19 –22 It could be envisaged that intervention with these DMTs could result in a legacy effect on disease activity, at least over the relatively short observation period of 12 months, as defined by the TERMS protocol. Interestingly, a recent study on the discontinuation of interferon beta therapy in subjects with high pre-treatment disease activity showed a relatively rapid return in relapse rate in this cohort. 23
Of note, those subjects on the TERMS study in the prospectively identified most active disease subgroup, ARR > 1, seemed to experience clinical benefit in response to Tovaxin® therapy over placebo regardless of DMT treatment history. The data suggest that disease severity prior to DMT may impact the duration of the legacy effect on DMT removal and entry into follow-on protocols. Taken together, prior treatment with DMTs may influence the ARR in the placebo group, and may in part explain the overall decline in ARR for placebo controls in more recent clinical trials. 24 Therefore, a decline in clinical activity in placebo controls will inevitably require larger sample sizes or extended periods of follow up for those studies to reach clinically significant endpoints.
Although clinical improvement, as defined by ARR, was noted in various sub-populations within the TERMS trial, including those naïve to DMT and those with high baseline ARR, no differences between treatment and control groups were seen for radiological endpoints. The presence of gadolinium enhancing (Gd+) lesions indicates areas of disruption within the blood brain barrier, and identifies sites more amenable to mononuclear cell recruitment. The mechanism of action proposed for Tovaxin® is the induction of regulatory T-cells that can target activated pathogenic MRTC. 11,12 Analysis of both inflammatory and regulatory T-cells has shown that both populations display the same potential to be recruited to sites of endothelial cell activation. In particular, lymphocyte homing is dependent on the expression of CD54 by both subsets of T-cells. 25 Therefore, it can be hypothesized that Gd+ lesions in Tovaxin®-treated subjects may be selectively enriched in T-regulatory cells that inhibit neurodegeneration in the target lesion, slowing progression of lesions to black holes and brain atrophy progression. The failure to detect significant differences in radiologic progression in the TERMS study may have resulted from the duration of observation being limited to 12 months.
Although this would be ideal, it is not possible to perform a detailed analysis of subsets of lymphocytes in the CNS lesions of subjects with RR-MS to find evidence to support the proposed hypothesis. However, evidence can be gleaned from pre-clinical animal models. For instance, studies in experimental allergic encephalomyelitis (EAE), a model of MS which typically requires pre-treatment of the animal with pertussis toxin to induce a breakdown in the blood brain barrier, have shown that EAE is amenable to therapeutic intervention without a requirement to block T-cell accumulation in the CNS. In particular, mice adoptively transferred with splenocytes from animals treated with glatiramer acetate, a registered drug for the treatment of MS, can prevent the development of clinical symptoms of EAE despite damage to the blood brain barrier and the accumulation of T-cells in the CNS. Analysis of the infiltrate showed the presence of T-cells with an anti-inflammatory profile, namely positive for transforming growth factor (TGF)b and IL-10. In addition, these cells were associated with the secretion of brain derived neurotrophic factor (BDNF), a mediator of neuroprotection. Most importantly, anti-inflammatory and neuroprotective mechanisms predominated, preventing the accumulation of overt clinical symptoms induced by priming animals with myelin antigen in adjuvant. 26 –28 The ability of Tovaxin® immunotherapy to induce a similar neuroprotective effect is an attractive hypothesis to account for the observed potentially contradictory radiological and clinical data observed in Tovaxin® treated subjects as defined by Gd enhancement.
In conclusion, some evidence of clinical efficacy in favor of Tovaxin®, as defined by ARR, was noted during the analysis of subgroups naïve to prior DMTs, as defined in post-hoc analyses. This suggests that previous DMT therapy can result in a legacy effect which may reduce study power to detect a meaningful treatment outcome if one is present. Small proof-of-concept studies may be selectively vulnerable to the risk of a Type II error. Therefore, the utilization of a cohort of treatment-naïve MS subjects may represent an important target population for early trials of candidate MS therapies. To increase the likelihood of detecting a clinically meaningful benefit in trials of short duration (12 months), studies in MS subjects with more active disease are also advised.
Footnotes
Acknowledgements
We extend our thanks to the research, development, GMP, operations, quality control, quality assurance, and clinical teams at Opexa Therapeutics for all of their efforts. The authors also would like to thank PharmaNet and Inventiv Clinical for performing the statistical analysis. We also acknowledge Shannon Inman and Don Healey, PhD, at Opexa Therapeutics for able project management of the study and for input on this manuscript, respectively.
This research was funded by Opexa Therapeutics, Inc.
Dr. Fox has received personal compensation for consulting services from Opexa Therapeutics.
Donna Rill holds stock options in Opexa Therapeutics as a part of her current employment compensation package.
Dr. McGuire has received personal compensation for consulting services from Opexa Therapeutics.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.