
Abstract
Keywords
Introduction
Multiple sclerosis (MS), a common disabling neurological disorder, has a prevalence between 50 and 200 per 100,000. 1,2 It is a chronic inflammatory demyelinating disease of the central nervous system (CNS) manifested morphologically by inflammation, demyelination, axonal loss and gliosis, 3 leading to a wide range of functional impairments. 4 Spasticity (muscle stiffness) is a common symptom of MS, occurring in more than 60% of people with MS. 5,6 It is a complex condition with negative effects including reduction in mobility, often making transfers more difficult. It can also be associated with painful muscular spasms and weakness, and predisposes to the development of contractures. 5 Conversely, however, some patients can find their spasticity helpful as they can use the stiffness to assist them with various aspects of their mobility such as walking or transferring. As the disease progresses, so does the spasticity, resulting in muscle spasms, immobility, disturbed sleep and pain. 7 Spasticity also interferes with patients’ ability to complete daily activities. 1 Disability resulting from spasticity can lead to patients requiring extensive nursing care. 7
Spasticity can be characterized as a ‘velocity-dependent increase in tonic stretch reflexes resulting from abnormal intra-spinal processing of primary afferent input’. 8 The Ashworth Scale was developed to assess this element of spasticity. However, following an upper motor neurone (UMN) lesion a person will present with a combination of sensorimotor signs and symptoms that are broadly classified as negative phenomena (which are normally characterized by a reduction in voluntary motor activity) and positive phenomena (which are normally characterized by increased levels of involuntary motor activity). 9 Thus, the term spasticity can also be used more generally to refer to the totality of the abnormal movement control caused by an UMN lesion, 10 and spasticity may be considered more as a collection of motor programme disturbances rather than a singlepathophysiological entity. 10 Spasticity, as defined by Lance 11 , is only one of the positive phenomena that may occur following an UMN lesion, 9 and other important aspects cannot adequately be assessed by the use of the Ashworth Scale.
Managing spasticity involves striking a balance between maximizing the beneficial effects whilst minimizing negative aspects of treatment. Current medication for spasticity includes baclofen, tizanidine, dantrolene, benzodiazepines and anticonvulsants. 5,7 Such therapies are associated with dose-limiting adverse effects, and there is a lack of good clinical data on which to judge their clinical effectiveness. Despite the widespread use of these oral anti-spasticity agents, many people with MS continue to experience moderately severe spasticity. 7 There is a clear need for new therapeutic agents to treat spasticity, as well as a need for assessment scales which reflect patients’ daily experience of their spasticity. 5
Cannabis sativa L. contains 60 or more cannabinoids, the most abundant of which are delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD). 12 Both of these have a pharmacology which suggests they may be useful in the relief of spasticity. 13 The endogenous cannabinoids (e.g. anandamide, 2-arachidonoyl glycerol [2-AG] and possibly others) act primarily via specific cannabinoid receptors: CB1 receptors, predominantly distributed in the CNS and CB2 receptors, located in both the CNS and extensively in the periphery (especially the immune system). 14 Both endogenous and exogenous cannabinoids have been shown to have an anti-spasticity effect in the recognized animal model of MS spasticity. 13
THC is a partial CB1 receptor agonist with principal pharmacological effects including analgesia, muscle relaxation, anti-emesis, appetite stimulation and psychoactivity. 14 CBD has anticonvulsant, muscle relaxant, anxiolytic, neuroprotective, antioxidant and antipsychotic activity and has been shown to reduce the anxiogenic and psychoactive effects of THC. 14-16
Sativex (USAN: nabiximols) (GW Pharma Ltd, UK), an extract of Cannabis sativa L contains THC + CBD at a fixed ratio, delivered as an oromucosal spray, each 100 µl of which delivers 2.7 mg of THC and 2.5 mg of CBD. Previously reported studies of up to 16 weeks’ treatment duration with Sativex showed significant improvement in the subject-reported severity of spasticity in MS. 17,18 In addition, a meta-analysis of three Sativex studies demonstrated benefit in this indication, 19 using a validated 0–10 Numerical Rating Scale (NRS). 20,21 A 20% improvement from baseline in the subject-reported spasticity severity has been shown to be the minimum clinically important difference, with a 30% improvement representing ‘much improved’. 20
A previous open-label long-term study suggests subjects with MS who derive symptom relief from Sativex in the first 10 weeks generally maintain that relief over an extended period of treatment without an increase in dose or an experience of intoxication. 22 When treatment was stopped suddenly in a cohort of subjects, no withdrawal syndrome was observed. 22
In this study we investigate the effect of the randomized withdrawal of Sativex in subjects who had been receiving benefit from its long-term use. This design not only allows assessment of the maintenance of efficacy in a placebo-controlled setting, but also allows for the more systematic assessment of any withdrawal syndrome.
Methods
This was a 5-week multicentre study (1 week baseline and 4 weeks treatment) conducted in five UK sites, to evaluate the maintenance of effect of Sativex in subjects with symptoms of spasticity due to MS who had been receiving long-term benefit from treatment (Figure 1). The study was approved by the relevant Ethical Committees and was conducted according to Good Clinical Practice guidelines.
Following informed consent and screening, eligible subjects entered a 7-day baseline period. During this baseline period, subjects were required to continue stable dosing with Sativex at their current effective dose level and complete a diary recording their daily spasticity severity and sleep disruption ratings using a 10-point NRS, and daily dosing information. At the end of this 7-day baseline period, subjects returned to the study site for Visit 2 (Day 1), ceased open-label treatment with Sativex and were randomized to either Sativex or placebo. They were asked to continue stable dosing at their current effective dose as identified at Visit 1. Subjects then attended an end of study visit at Week 4 (Visit 3, Day 28) or earlier if they withdrew from treatment. Spasticity and sleep disruption review and dosing diaries were completed each day during the open-label and randomized treatment periods.
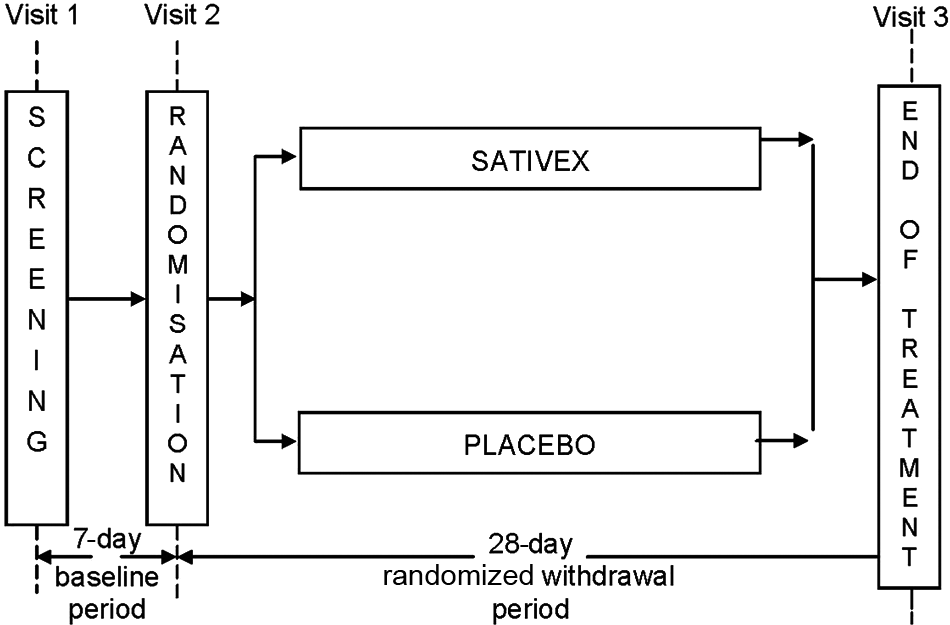
Study design
Inclusion and exclusion criteria
Study entry inclusion criteria
Subjects diagnosed with MS and receiving Sativex for the relief of spasticity for at least 12 weeks prior to screening, and who were judged to have been receiving benefit from and showing tolerability to Sativex, were eligible for inclusion in the study. Subjects taking other medications affecting spasticity were required to have remained at a stable dose for at least 3 months prior to study entry and be willing to maintain this for the duration of the study.
Study exclusion criteria
Any subjects who had a concomitant disease or disorder that had spasticity-like symptoms or that may have influenced the subject’s level of spasticity were excluded. If subjects were unable to rate their level of spasticity or distinguish it from other MS symptoms, they were excluded. Any subjects who had received botulinum toxin or rimonabant, a cannabinoid receptor antagonist, in the 3 months prior to study entry were also excluded. Any subject with a concurrent history of significant psychiatric, renal, hepatic, cardiovascular or convulsive disorders was also excluded, as were subjects with a known or suspected history of alcohol or substance abuse.
Study endpoints
Primary endpoint
The primary efficacy endpoint was the time to treatment failure (TTF), with the null hypothesis that the TTF would be the same in subjects randomized either to placebo or to Sativex. Treatment failure was defined as:
a) cessation of randomized treatment before Visit 3 (i.e. subjects who withdrew from the study without completing the Day 28 visit)
OR
b) a worsening of spasticity (defined as an increase in the mean spasticity NRS over the last seven days of the treatment period of at least 20% and at least one unit from the treatment baseline)
OR
c) a clinically relevant increase in or addition to anti-spasticity medicines or disease-modifying medications after randomization.
Secondary endpoints
A range of secondary measures were also assessed:
Subject Global Impression of Change (SGIC)
daily spasticity severity NRS score
the daily sleep disruption NRS score
Modified Ashworth Scale score
Motricity Index.
Functional assessments included the Carer Global Impression of Change (CGIC) and a timed 10-metre walk.
Safety endpoints
The safety endpoints were adverse events (AEs), laboratory parameters, vital signs, oral and physical examination.
Statistical methods
Sample size
It was estimated that approximately 60 subjects (30 per group) would be willing to enrol into this study from a database of patients that were using Sativex on a Named Patient Supply basis. There were no data available upon which to make assumptions with regard to the rate of deterioration in spasticity once Sativex treatment was withdrawn, and so there was no information with which to base an estimate of the hazard ratio for treatment failure. Consequently, it was not possible to adequately justify any pre-specified sample size calculation. In addition, it was expected that there would be significant difficulty in recruiting into this study, with a very limited population of eligible subjects, and so the possibility of recruiting subjects into a pilot trial to obtain the information needed to calculate the sample size was not a feasible option. Thus, a required estimated sample size of 60 subjects was specified for the study (n = 30 per group), assuming that this target was the maximum achievable and would provide useful information on maintenance of effect.
Randomization
Following informed consent and screening, eligible subjects were provided with and asked to commence treatment with Sativex at their current effective dose level during the baseline period. At Visit 2, after continued eligibility had been confirmed, subjects were randomly allocated to one of the two treatment groups. An independent statistician produced a treatment allocation schedule using balanced randomly permuted blocks of 4 using a computer-based algorithm.
Primary endpoint
The primary endpoint of the TTF in the randomized withdrawal phase was summarized and analysed using the Randomized Withdrawal Analysis Set. This was analysed using Kaplan–Meier survival analysis methodology and Cox Proportional Hazard Regression.
The Kaplan–Meier analysis was used to provide estimates of treatment failure by time (treatment failure was defined as either cessation of treatment, or a 20% increase in spasticity or taking additional anti-spasticity medication). Proportional hazards modelling was used to compare the treatment arms and provided a hazard ratio (HR) for the hazard of failing treatment within each arm. The proportional hazards model had ‘TTF or censoring’ as the dependent variable – data were censored upon study completion with no treatment failure – and the model included randomized withdrawal treatment as factor. The proportional hazards regression method modelled the TTF.
All of the secondary endpoints were predefined, although the p-values have not been corrected for multiplicity and so care should be exercised in their interpretation. The change in spasticity severity NRS, sleep disruption NRS, Modified Ashworth Scale, Motricity Index and timed 10-metre walk from baseline to end of study were analysed using an Analysis of Co-Variance (ANCOVA) and had the baseline assessment of the endpoint as a covariate and treatment as factor in the model. Both the SGIC and CGIC were analysed with ordinal logistic regression using the cumulative proportional odds model, with treatment as the only factor in the model.
All statistical comparisons between treatments used two-sided statistical tests at an alpha level of 10%. This alpha level was selected when designing the study in an attempt to keep the confidence intervals (CIs) narrow enough to be of value, given it had not been possible to estimate the power of the study.
Results
A total of 37 subjects were screened, of whom only 36 could be recruited. Many subjects were very concerned about the possible return of their symptoms even though they were assured that they could drop out of the study at any time. Of the 37 subjects screened, one failed to meet the entry criteria (Figure 2) thus 36 subjects entered the randomized withdrawal period; 18 subjects received Sativex and 18 received placebo.
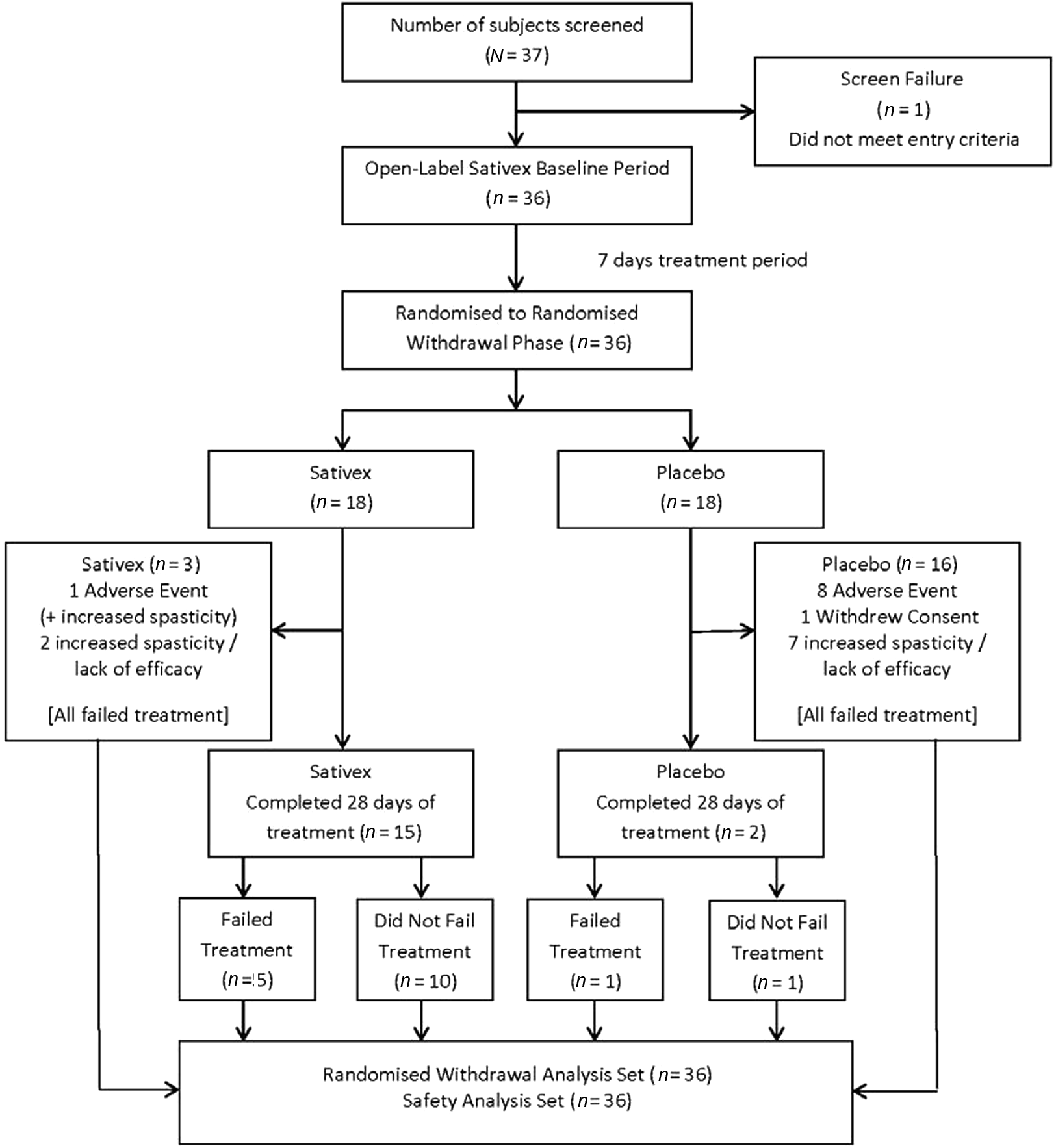
Disposition of subjects
Study population demographics are presented in Table 1. The two treatment groups were well matched for age, gender, duration and type of MS (mean >16 years) and Expanded Disability Status Scale (EDSS) score (7.0). The mean age of the subjects was 57 years and all were white Caucasians, with a mean baseline spasticity severity score (NRS) of 3.6 (SD 1.7) in the Sativex group and 4.13 (SD 2.2) in the placebo group. The active treatment group had been using Sativex for a mean duration of 4.2 years (median 2.0 years) and the placebo group for a mean of 3.0 years (median 1.9 years), and were taking a mean daily dose of 7.33 (median 6.5 sprays) and 9.17 sprays (median 6.0 sprays), respectively. A total of 21 subjects (58%) had an EDSS score of 7.0 or more, and were therefore classified as non-ambulatory.
Demographics and baseline characteristics – Randomized-withdrawal period
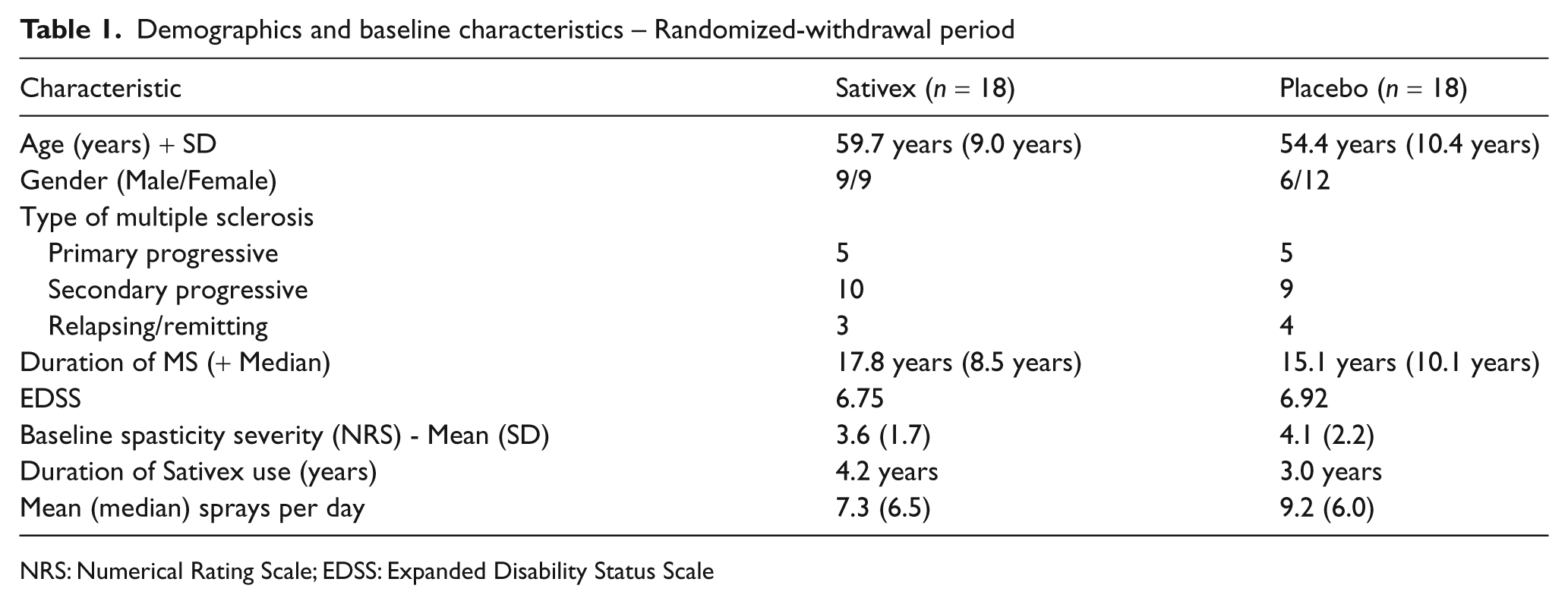
NRS: Numerical Rating Scale; EDSS: Expanded Disability Status Scale
Concomitant medication
In total, 22 subjects (61%) were taking other anti-spasticity medication at the start of the study, with baclofen being the most common. The other anti-spasticity agents used were benzodiazepines (16%), other analgesics and antipyretics (i.e. gabapentin, 16%), quinine and derivatives (quinine sulphate, 12%), other antiepileptics (pregabalin, 3%), adamantane derivatives (amantadine hydrochloride, 3%) and 3% on herbal preparations. There were no apparent differences between the treatment groups in terms of concomitant medications taken by the groups. Four placebo patients were taking benzodiazepines during the randomized withdrawal period, compared with one subject on Sativex. Similarly, nine placebo patients were taking centrally acting agents (tizanidine, baclofen) during the randomized withdrawal period, compared with six subjects on Sativex.
During the randomized withdrawal study period, five subjects (14%) were taking disease-modifying medications at the start. One placebo subject started taking steroids for an acute flare up of MS after her GP prescribed them to her. Thus, a total of six subjects (17%), three from each treatment group, were taking disease-modifying medications during the randomized period.
Primary endpoint: TTF
Of the 36 subjects randomized, 17 completed the 4-week treatment period (15 in the Sativex group and two in the placebo group), with the other 19 withdrawing from the study. Some of the subjects who completed the 4-week treatment period, however, met the treatment failure criteria (Sativex n = 5, placebo n = 1). By the end of the 4-week randomized withdrawal period, 17 of 18 (94%) subjects from the placebo group had failed treatment compared with eight of 18 (44%) subjects from the Sativex group.
Six subjects receiving placebo failed treatment with the primary reason for failure being due to cessation of study medication before Visit 3 (Week 4)
Eight subjects receiving Sativex and 11 subjects on placebo failed treatment with the primary reason for failure being due to a worsening of their spasticity (i.e. ≥ 20% increase in spasticity); i.e. the spasticity worsened before they stopped study medication
No subjects failed treatment due to a clinically relevant increase in anti-spasticity medication or disease-modifying medication after randomization.
The TTF was calculated as the number of days from the first day of randomized treatment up to the first day of treatment failure. The first day of treatment failure was the earliest of:
The day of premature cessation of randomized study medication
The earliest (or first) day for which the mean spasticity NRS from that day through to the end of treatment had worsened by at least 20% and at least 1 unit from the baseline score.
TTF was analysed as a HR, where 1 would indicate an equivalent outcome for Sativex and placebo. This analysis was statistically significant in favour of Sativex, with the hazard of treatment failure for subjects on placebo being three times that of those receiving Sativex (HR = 0.335, 90% CI: 0.162, 0.691; p = 0.013). The Kaplan–Meier type plot of the data is shown in Figure 3.
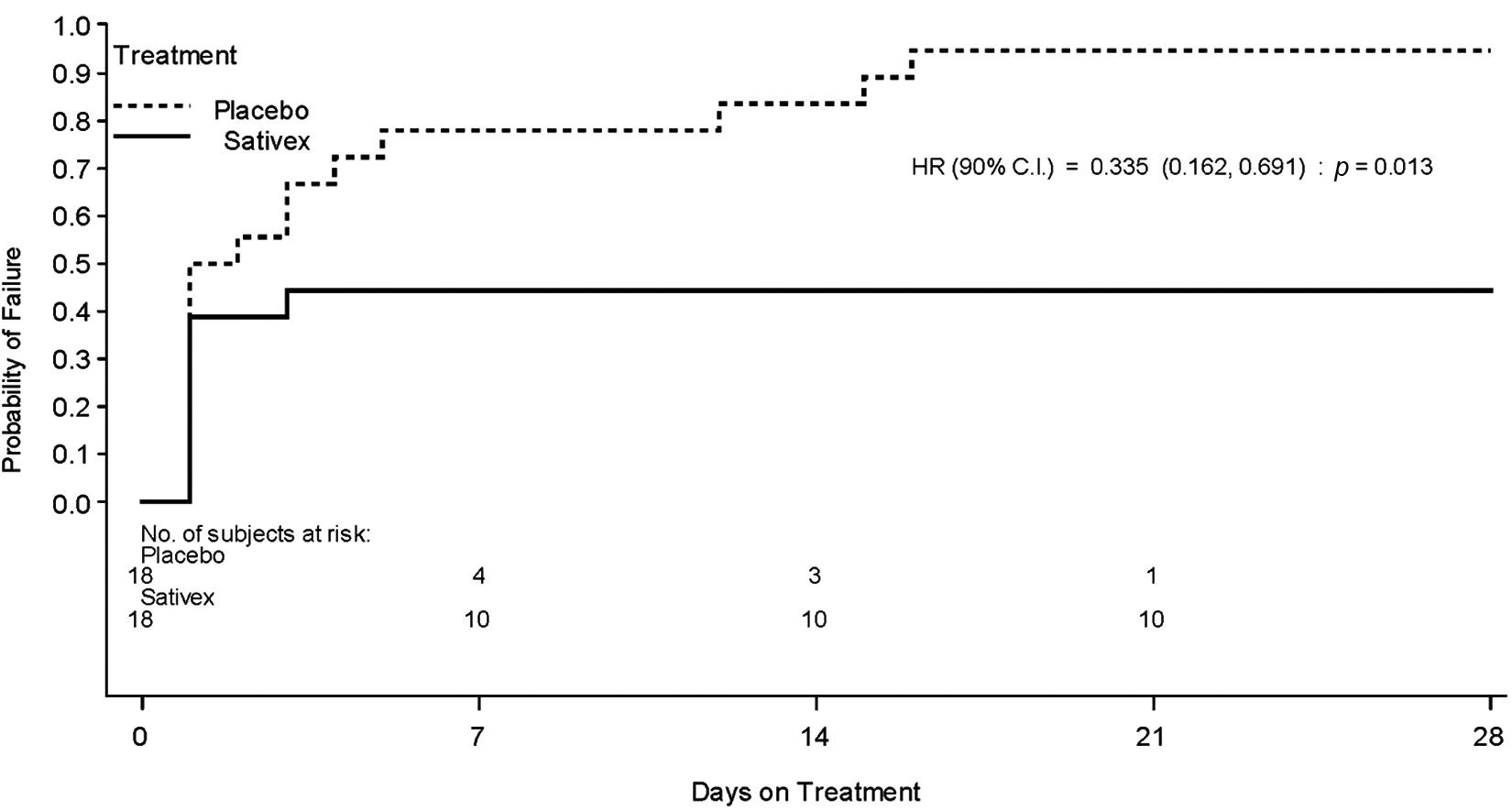
Kaplan–Meier Plot showing the time to treatment failure for subjects
A summary of the treatment failure status of each subject is presented in Table 2. The median TTF in the Sativex group was more than 28.00 days), compared with a median of 1.50 days in placebo group).
Summary of time to treatment failure by subject (with reason for failure)
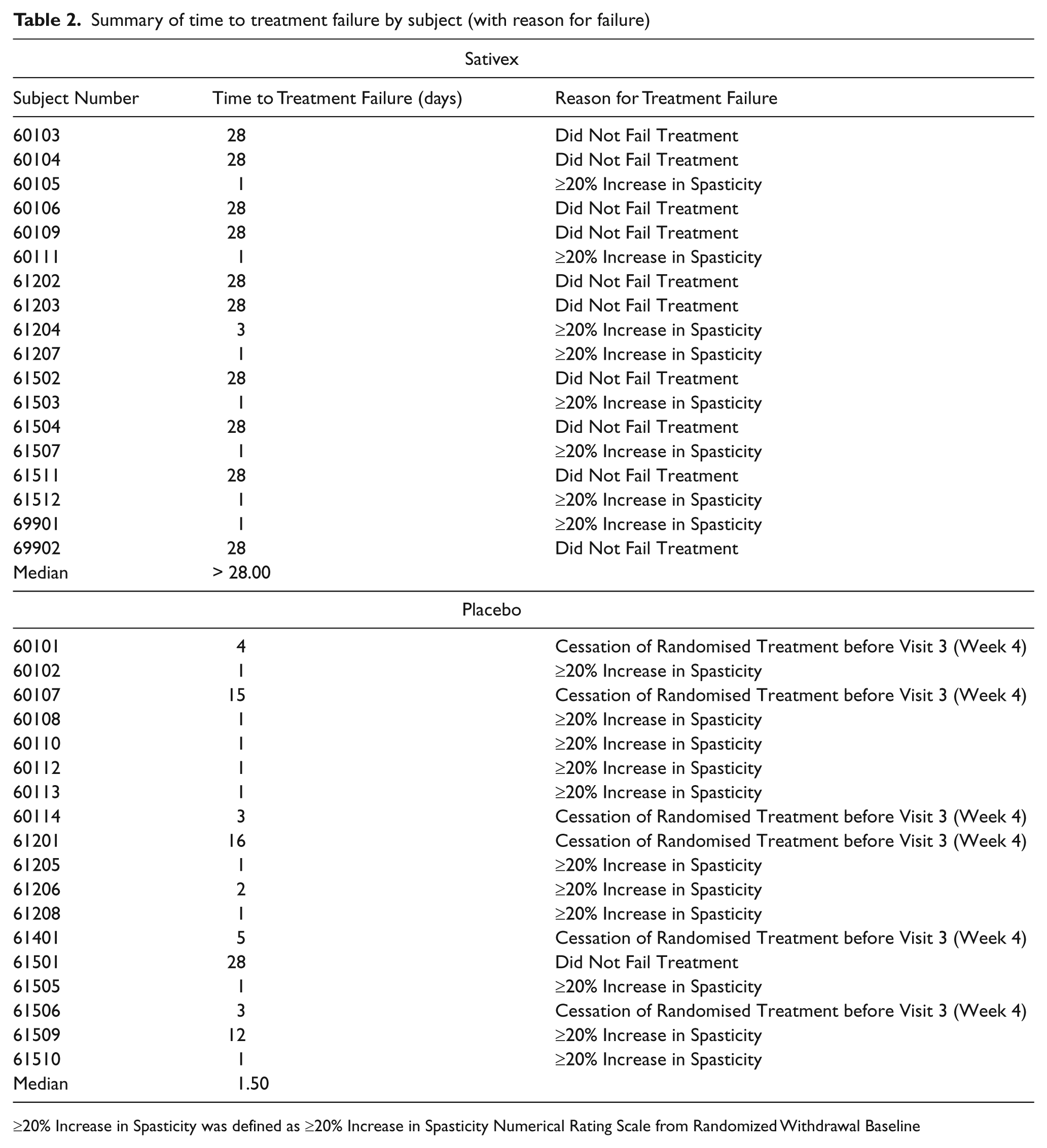
≥20% Increase in Spasticity was defined as ≥20% Increase in Spasticity Numerical Rating Scale from Randomized Withdrawal Baseline
Secondary endpoints
Spasticity NRS score
Sativex subjects showed an increase (deterioration) in adjusted mean spasticity NRS of 1.00 point from a mean baseline score of 3.60 points, compared with an increase of 1.21 points from a baseline of 4.13 points for placebo. The estimated treatment difference of -0.21 points (90% CI: -1.22, 0.79 points) was not statistically significant (p = 0.720).
Sleep disruption NRS score
Subjects receiving Sativex showed a deterioration in quality of sleep of 0.60 points from a mean baseline score of 2.31 points, compared with a deterioration of 1.24 points from a baseline of 2.11 points for subjects receiving placebo. The estimated treatment difference of 0.64 points (90% CI: -1.60, 0.33 points) was not statistically significant (p = 0.271).
Modified Ashworth Scale
For subjects receiving Sativex, the adjusted mean change in the Modified Ashworth Scale was 1.11 points compared with a change of 1.64 points for placebo, from baseline scores of 23.2 and 23.0 points, respectively. The estimated treatment difference of 0.53 points (90% CI: -4.68, 5.74 point), was not statistically significant (p = 0.862).
Timed 10-metre walk
Ambulatory subjects in the Sativex group experienced a deterioration in their adjusted mean walk time of 3.46 s (from a mean baseline score of 40.1 s) compared with a deterioration of 5.24 s (from a baseline of 24.1 s ) for those subjects receiving placebo. The estimated treatment difference of 1.78 seconds (90% CI: -14.52, 10.96 s), was not statistically significant (p = 0.808).
Motricity Index
The Motricity Index was completed for affected arms and legs by one and four subjects, respectively, in the Sativex treatment group, and seven and 16 subjects in the placebo group. No significant differences between treatments were found for the leg assessments (p = 0.300).There were too few subjects to make any form of comparison in the arm assessment; the data are consistent between the treatment groups.
As subjects who withdrew early from treatment were not always assessed immediately on withdrawal (but were allowed to re-start their Sativex if they withdrew from the study), some subjects had started back on their own supply of Sativex before they returned to the unit for their formal withdrawal visit. The interval between day of cessation of study medication and study visit varied between withdrawn subjects. Blinding was maintained throughout and beyond the study, even when patients returned to the study following cessation of study medication.
Due to patients immediately being able to revert to their previous long-term Sativex treatment upon withdrawal from study, the numbers providing data to the secondary outcome measures were less than the total population. Also, the number of ambulatory subjects was small, and so walking time assessments included only those patients able to complete the 10-metre walk. For these reasons, no meaningful conclusions can be drawn from these endpoints.
SGIC
The odds of being improved (i.e. in a better category) were greater in the Sativex than the placebo group, with an odds ratio of 4.55, which was statistically significant (p = 0.017; 90% CI: 1.59, 14.00).
At the end of the study, six of 18 subjects (33%) receiving Sativex described their symptoms in the category of ‘no change’ compared with one (6%) subject on placebo. Twelve of 18 subjects (67%) on placebo and six of 18 subjects on Sativex (33%) described their symptoms as ‘Much Worse’ or ‘Very Much Worse’.
CGIC General functional abilities
Of the 36 carers of subjects from both treatment groups, a total of 10 (Sativex group) and 14 (placebo group), completed the CGIC. All 14 carers of subjects who received placebo categorized the subject’s functional ability as worsening, compared with 70% of the carers (7/10) of subjects who received Sativex. The odds of being improved for the Sativex group were statistically significantly higher than for the placebo group (odds ratio 18.55; 90% CI: 3.94, 118.77; p = 0.001).
CGIC Ease of transfer
A greater proportion of the carers of subjects in the placebo group than in the Sativex group categorized the subject’s ease of transfer as worsening (Sativex, 70% and placebo, 86%). The odds of being improved were higher for the Sativex than the placebo group (odds ratio 3.44; 90% CI: 0.95, 13.72; p = 0.115).
Safety and tolerability
The mean daily dose of Sativex was 7.7 sprays (median 6.4) compared with 9.0 sprays (median 6.0) of placebo. The median number of days actual exposure in the Sativex group (28.5 days) was approximately five times greater than in the placebo group (5.5 days).
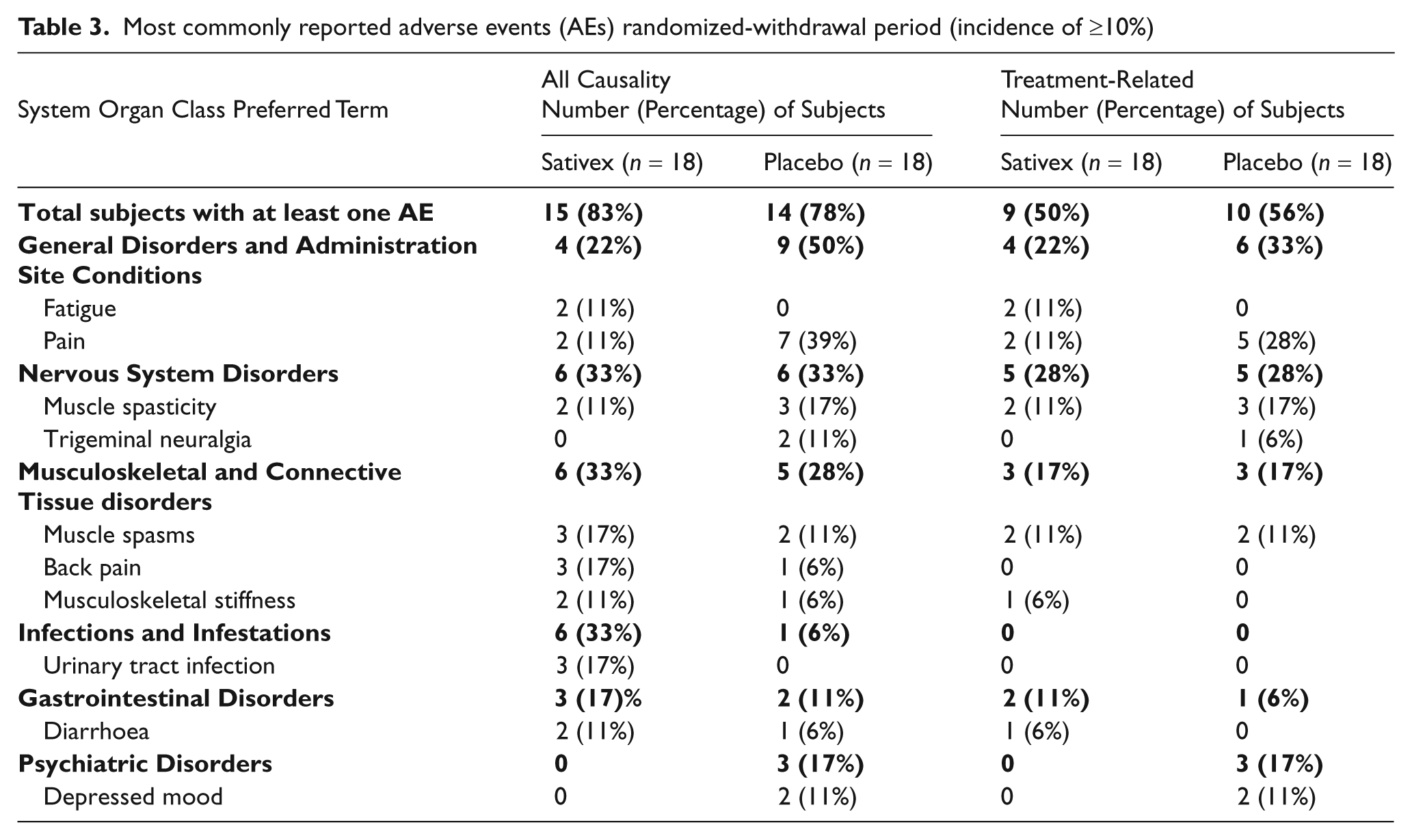
During the placebo-controlled randomized withdrawal period, 15 of the 18 subjects (83%) receiving Sativex and 14 of the 18 subjects (78%) receiving placebo experienced a treatment-emergent AE. The most common treatment-related AEs were pain (seven subjects: two (11%) receiving Sativex; five (28%) receiving placebo), muscle spasticity (five subjects: two (11%) receiving Sativex; three (17%) receiving placebo), muscle spasms (four subjects: two (11%) in each of the Sativex; and placebo groups) and depressed mood (two subjects: both (11%) receiving placebo).
The majority of AEs were considered mild or moderate. During the randomized withdrawal period, only four subjects (two in each treatment group) experienced a severe AE. No deaths were reported during the study. There was only one serious adverse event (SAE) (pain in hip and thigh and lumbar spinal stenosis) in a subject randomized to Sativex, which was considered unrelated to study medication.
The only AE reported in association with abnormal laboratory values was a mild non-serious increase in GGT in a subject receiving Sativex.
Most commonly reported adverse events (AEs) randomized-withdrawal period (incidence of ≥10%)
Discussion
The Enriched Enrolment Randomized Withdrawal study design (EERW) is recognized as being able to provide reliable evidence of long-term efficacy, in particular in conditions where long-term treatment with placebo may not be acceptable. 23 One of the virtues of the design is that exposure of patients to placebo can be short, yet it still allows for an assessment of long-term efficacy. In this setting, the demonstration of efficacy depends upon the occurrence of treatment failure in significantly more subjects exposed to placebo compared with the active treatment. Since the randomized withdrawal study design depends on identification of subjects who are failing on their assigned treatment, the primary endpoint must take this into account. In this study, we used the time to treatment withdrawal to meet this design need. The concept of therapeutic failure as a primary endpoint is one that has been accepted by regulatory authorities for some years, 24,25 and also resonates with the way that medicines are used in clinical practice. A prescriber is unlikely to keep a patient on a medication which is failing to provide benefit. One of the criteria used to judge treatment failure was the need for rescue medication. In this way, the EERW study can also incorporate valuable features of other study designs. Recruitment into this type of study can be difficult, and a number of prospective subjects declined participation as they did not wish to run the risk of stopping a treatment which had proved successful for them and thereby re-experiencing the unpleasantness of their symptoms. This limited the total availability of subjects.
Because of the small sample size and the lack of a formal sample size calculation, one should exercise caution in the interpretation of the significance of this result. Nonetheless, this (EERW) placebo-controlled study confirms that the efficacy seen in short-term studies of Sativex in people with spasticity due to MS appears to be maintained in long-term use and provides support to previously published open-label studies of Sativex. 22,26 The primary efficacy endpoint, the TTF, was statistically significant in favour of Sativex (p = 0.013; HR = 0.335; 90% CI: 0.162, 0.691). Sativex was already being used as an add-on therapy to gain adequate relief in subjects with advanced MS, and the majority were already on currently available anti-spasticity medication. All of the subjects entered into the study were judged to have been receiving benefit from and showing tolerability to Sativex, in both the investigators’ and subjects’ opinion.
Of the secondary endpoints, only the SGIC and CGIC achieved significance. However, this is not surprising in view of the small subject population with a high EDSS (58% > 6.5). The significant difference between Sativex and placebo for the SGIC (p = 0.017) and the CGIC (functional ability) (p = 0.001) is of note because it shows that the carer was able to distinguish the difference in the change in the level of function between those patients who continued active treatment and those who were randomized to placebo.
The lack of significant difference between treatments with regard to the mean spasticity NRS scores is unsurprising, not only due to the small number of subjects enrolled in this study (i.e. limited power of the study) but is also probably due to the make-up of the study population – a group of patients who had previously been responding well to treatment with Sativex on a long-term basis (mean = 3.6 years of Sativex exposure). Some 44% of the Sativex subjects reported deterioration in their spasticity whilst on study; such a worsening of spasticity in almost half of an already small active-treated population will undoubtedly have diluted the mean spasticity NRS scores for the overall Sativex group. Despite this, the mean spasticity NRS response was still in favour of Sativex when compared with placebo. This suggests that there is a nocebo (reverse placebo) effect present in this study. It has been a longstanding clinical notion that an individual’s beliefs and expectations can significantly influence the therapeutic benefit and adverse effects of a pharmacological treatment. 27 Placebo and nocebo responses may be triggered by psychosocial variables forming the treatment context, such as expectation of treatment outcome via verbal cues, previous experience, or patient–physician interactions. 27
It should be noted that all patients who entered the study were aware that they had a 50% chance of being randomized to placebo. The anticipation of deterioration was probably responsible for subjects in the Sativex treatment group indicating that they had deteriorated, despite the fact they were continuing on the same dose of a medicine that they had been using successfully for years. Even though there was no formal testing of whether subjects remained blind to treatment allocation, this does suggest that they were not able to tell placebo from Sativex, and that unblinding was unlikely to have occurred.
Since this type of study design involves the sudden stopping of a long-term treatment, it carries the risk of precipitating a withdrawal syndrome. Such a syndrome has been described in heavy recreational cannabis users to the extent that there is now a recognized cannabis withdrawal syndrome. 28 The lack of such a syndrome when subjects with MS have been suddenly withdrawn from Sativex has been reported previously, in an open-label setting. 22 This small study showed no evidence of a withdrawal syndrome in subjects who stop Sativex suddenly, despite a prolonged period on the medicine. Overall, there was a similar frequency and severity of AEs in both the Sativex and placebo groups, with more than 85% of such events being deemed mild or moderate in severity. Although the number of subjects in this study was small, the presence of no new significant safety issues provides further support to previous studies.
These findings are important because until now, the evidence for long-term maintenance of efficacy of Sativex has come from long-term open-label exposure and clinical observation. While this is the means by which many physicians will reach a view of the long-term risks and benefits of a medicine in a ‘real world’ setting, the quality of evidence from such studies is not sufficient to allow for firm conclusions to be drawn. It has previously been reported that responders to Sativex may be easily identified by a simple trial of therapy. 29,30 This study therefore provides further evidence of the maintenance of long-term efficacy of Sativex as an add-on therapy in the treatment of MS spasticity, in patients who have already been identified as responders.
Footnotes
This study was funded by GW Pharma Ltd.
GW Pharma Ltd produces Sativex (nabiximols) and is licensed for the treatment of spasticity in multiple sclerosis in the UK. W Notcutt receives a research grant from GW Pharma Ltd. R Langford, P Davies and S Ratcliffe were all investigators on this study and received Investigator Fees for their participation in this study. R Potts is an employee of GW Pharma Ltd and holds equity in the company.