
Abstract
Background:
A review of Health Canada’s post-market surveillance database has revealed that the reporting of adverse events (AEs) following aesthetic injectable treatments is significantly underreported. To increase reporting, investigators have recently developed a novel Electronic Data Capture system: The Global Registry of Adverse Clinical Events (GRACE©).
Objective:
To identify the incidence of AEs associated with aesthetic injectable treatments.
Methods:
Aesthetic clinicians from 10 Canadian sites were recruited. Demographic and clinical data were recorded within the database, which included over 45 patient variables.
Results:
Throughout the active phase of the trial (duration: 27 months), 123,124 injectable treatments were conducted. One hundred and eleven patients, experiencing a total of 235 AEs, were entered into the portal. This equated to an AE incidence rate of 0.19%, per treatment. Thirty unique products were associated with AEs. In total, there were 112/235 (47.66%) mild, 88/235 (37.45%) moderate, and 35/235 (14.90%) severe AEs. The most common complication (n = 48/235; 20.43%) was swelling, with a prevalence of 0.04%. Of the documented AEs, only 5 were reported to other sources, including 1 case being reported to Health Canada and 4 cases to the respective product manufacturer.
Conclusions:
The initial feasibility of a registry assessing safety outcomes following injectable treatment has been demonstrated. Findings support that the implementation of the GRACE Portal is an effective outreach strategy for increasing AE reporting by health care professionals. The data represent a more accurate depiction of the safety profile of approved aesthetic injectables in Canada.
Introduction
The rates of adverse events (AEs) following injectable treatments are widely varied within the literature.1-13 Supplemental Material 1 displays the summary results of 14 studies reporting AE rates and demonstrates a widely variable range of rates depending on the source document. For example, a common AE following injection is erythema, and this has been reported to occur in as little as 0.004% to as high as 100% of treated patients.7,14 The disparity between reported prevalence rates may be due to many contributing factors. Some may include variations in the record-keeping practices of different clinics (eg, many may not record immediate local site reactions to the injection technique as AEs and instead only report late onset and/or chronic AEs) and insufficient follow-up with patients (eg, requiring that patients follow-up with the clinic may result in an underreporting of minor AEs and only allow for the practitioner to become aware of more stressing AEs experienced by patients). Moreover, the disparity between the prevalence observed during clinical examination and those documented suggests that AEs are being significantly underreported.15-18 For example, as transient local site reactions to injections are a part of the normal immune response to injury, 15 it seems intuitive that in the majority of patients, clinicians should observe one or a combination of any of the following: erythema, edema, inflammation, redness, and soreness. However, some sources report very rare occurrences of an acute response (eg, 0.06-0.15). 16 Providing a distinct overview of the specific symptoms experienced by patients also becomes less discernable depending on the language used in the literature. Oftentimes, authors do not report the individual rates of specific AEs (eg, erythema, edema) and instead report an overall rate for all “local site reactions” or “immune responses.”16-18 This contributes to the uncertainty regarding the exact prevalence rates of specific symptoms. Furthermore, given the large number of injectable treatments performed (ie, 17,139,392 injections in the United States in 2023), 19 it is evident from current databases that AEs are being underreported. For example, a search of the “Canada Vigilance Adverse Reaction Online Database” revealed that only 10 cases of AEs were reported for the millions of treatments performed. Furthermore, between 2010 and 2019, Health Canada only received 48 adverse incident reports of pain, edema, nodules, abscesses, lip necrosis, partial loss of vision, and vascular compromise suspected of being associated with the use of dermal fillers.20,21
Given the marked underreporting of AEs and the variation in rates currently reported, the establishment of an easily accessible online AEs registry could provide the observational platform required to collect and evaluate data on the incidence of AEs following injectable treatments. Furthermore, this AEs registry would also provide the preliminary data required to create algorithms or protocols for managing AEs and standardizing care. In consideration of the above, the present manuscript described the development and validation of an AE database.
Research Question
What is the true incidence of AEs associated with aesthetic injectable fillers, in routine clinical practice?
Objectives
To identify the incidence of AEs associated with aesthetic injectable treatments.
Methods
Portal Design and Development
Major milestones in the development of the AE registry are depicted in Supplemental Material 2. The portal was constructed specifically for observational research as a cost-effective management tool (differential product analysis on file). The design supports simple delivery models that are scalable, flexible, and have the potential for international dissemination. Moreover, the registry consists of a transparent, uncomplicated user-face (Supplemental Material 3). Data collection occurs through electronic clinical report forms (eCRFs), which are form-like web pages. Through eCRFs, sites submit patient, treatment, and AE data to the central database. The eCRFs have the same structure and design as typical paper-based CRFs from which their content was based. The eCRFs are organized into sections: (i) Patient Information; (ii) AE Information; and (iii) Follow-Up Visits. This structure introduces automation to the AE reporting process by providing a platform that intuitively breaks the procedure down into its primary steps. As the main outcome variables of interest are AEs, reporting is an event-centered process, rather than a visit centered one. A flexible visit structure ensures that users can individualize the number of follow-up visits and only have to enter data for visits that actually occur.
Throughout the data entry process, intuitive prompts help clinicians move swiftly through the entry procedures. Mouseovers provide definitions of terms to ensure consistency of imputed data across all sources. Individual sites are able to generate their own reports and data extracts, and the Super Admin (eg, study sponsor) can generate reports and data extracts for all sites. This feature eliminates data transcription and iterations and ensures that AE and product quality data capture is made easy.
A differential product analysis (data on file) demonstrated that at the time of development, there were no Electronic Data Capture (EDC) systems specifically designed for AE data capture. This fact illuminated many purpose-driven limitations to the available platforms. Therefore, the GRACE Portal platform includes a purpose-built system design (eg, risk-based remote monitoring, back-up, and disaster recovery) and efficient eCRF design (eg, proper data element identifiers, robust use of edit checks, and electronic prompts for missing or inconsistent data). These features eliminate a high percentage of possible errors from manual inputting and save money on monitoring travel expenses for source verification site visits. Upon entry of an AE, the system automatically creates and records a link to the appropriate International Statistical Classification of Diseases and Related Health Problems, 10th revision (ICD-10) code and term, to ensure consistent reporting between clinicians and sites. Also, this software is the first and only EDC to subsequently provide links between the reported AE and current, real-time guidelines to standards of treatment and consensus documents on how to address the AE. At the final visit, a page displays a summary of all entries for that patient. At this point, the clinicians can make any necessary changes; then, they will e-sign and date stamp the record to archive it. No further changes to the patient’s data can be made after this point unless the monitor issues a query.
Regulatory Requirements
Many features of the portal have been designed with certain international and national regulatory requirements in mind. For example, the registry design conforms to the following privacy and security standards:
Health Insurance Portability and Accountability Act (HIPAA): The Privacy Rule, or Standards for Privacy of Individually Identifiable Health Information, which establishes the national standards for the protection of certain health information; and The Security Rule, which establishes a national set of security standards for protecting certain health information that is held or transferred in electronic form.
FDA 21CFR Part 11(Code of Federal Regulations Title 21): States general provisions for electronic records and signatures (eg, contains e-signatures, data validation, access management).
ISO (The International Organization for Standar-dization) 9001:2015: States the requirements for a quality management system (eg, audit trial and corrective and preventive action).
Patient confidentiality
As the registry only required data from the injector(s) or site(s), the data were anonymized and de-identified at source. No individually identifiable health information or “protected health information” as defined by HIPAA and its implementing regulations were collected. After the initial patient file was created, the clinician kept adding data to the registry by using an automatically assigned subject number.
Data encryption
The platform was equipped with encryption capabilities (ie, AES256 NHS benchmark).
Secure Sockets Layer certificate
Secure Sockets Layer (SSL) certificates are small data files that digitally bind a cryptographic key to an organization’s details. When installed on a web server, it activates the padlock and the https protocol and allows secure connections from a web server to a browser. The software utilizes SSL certificates to secure data transfer and logins.
Risk plan for ensuring data quality
Standard operating procedures ensured that site quality checks were routinely performed to reveal information such as time since last patient entry, open queries, and total queries. “Delinquent” sites were first put on a watch list that will trigger a call from the Sponsor. In the event that delinquency continued, a site visit would be justified to further train staff or review source documents and speak with the investigator to resolve any issues. This stepwise process better allocated resources to delinquent sites.
AE coding and terminology
The platform was equipped with a medical dictionary for AE coding and terminology. This feature improved the dissemination potential of research conducted using this software and also simplifies and quickens the task of reporting AEs to regulatory bodies. The system includes pre-coded colloquial terms, which map back to the appropriate ICD-10 terms and codes.
Different operational definitions may affect incidence rates, and in general, there is a low-level consensus on which operationalizations are best. For analyzing data captured in administrative databases or electronic health records, the incidence of patients with at least one encounter with a healthcare professional (HCP) for an AE during the evaluation period is often used. 22 Therefore, in the present study, HCP-recorded diagnostic information was used for determining the number of incident cases.
Technical requirements
The system is accessible online via Chrome and Safari web browsers. Providing a web-based solution (in comparison to a desktop application) circumvents the installation process and expedites updates to software for users. The portal is available on Windows and Mac.
Given that this EDC software is a cloud-based system, it lives on the internet (unlike on-premise solutions). This allows flexibility as the central server can be located anywhere in the world and the software can be accessible to anyone with internet access. This makes the system more functional, affordable (since the same software base code can be used by all customers), and allows for more frequent updates. There is no need for customers to build and maintain an on-premise IT infrastructure that requires data centers, real estate to house them, or skilled professionals to operate them. Also, the fact that very little user training is required, and that training can be done remotely, eases and minimizes the costs associated with the process of initial setup, and facilitates penetration into various graphical regions.
Moreover, the GRACE Portal contains some exclusive and uncommon system features, including:
Being the only system that provides guidance and consensus information upon AE reporting.
Data encryption of identifying and protected health information.
Contains an uploading tool for any associated images (available when confirmation that a signed photo release form has been obtained) or documents (eg, lab results) that may need to be attached to individual records.
Flexible visit structure, which is a feature many other EDC programs do not offer—which triggers many queries or missing data fields and causes problems during data analyses.
Pre-coded colloquial terms within the system—easy terminology which maps back to appropriate ICD-10 terms.
Ease of use and simple interface: existing software may be difficult to support technically or the creation of study-specific eCRFs may require advanced programming and coding knowledge.
Portal Validation
User acceptance testing
After completing the design and development of the GRACE portal, we approached colleagues to engage in user acceptance testing. These included evaluations related to the likeability of the user-face, its ease of use, the need to include or exclude certain variables, and the phrasing and interpretations of questions.
Pilot study
Aesthetic clinicians from 3 Quebec clinics were invited to participate in the registry’s validation. Over a 3-month period, clinicians were asked to report all AEs related to neurotoxins or soft tissue fillers. Variables of interest included sociodemographic data, product type, AE location, duration, intensity, and outcome. Data quality was evaluated by assessing completeness and accuracy (defined as internal consistency within the registry record and alignment with the patient’s source medical record). After revisions based on user feedback, Version 5.0 of the registry was deployed as the final version used in the pan-Canada study.
Study Design
Plastic surgeons and dermatologists were invited to participate in the registry. The clinicians were asked to report any AEs related to filler injections that occurred throughout the thirty-month data collection period. The study protocol included a description of how AEs were to be classified by standard definitions (ie, for severity, expectedness, and potential relatedness to treatment). The proposed registry focused on information regarding AEs obtained from observational methods. All AEs related to the use of soft tissue fillers for aesthetic purposes in the face and neck area were included. There were no other patient-related inclusion or exclusion criteria. Assessments included clinical events (eg, vascular occlusion and skin necrosis) and patient symptoms (eg, pain). Personal data, prior injectable filler treatments, reaction type, location, duration, intensity, and outcome were also recorded.
Recruitment consisted of all patients presenting with an AE following treatment with injectable products. The duration of treatment and number of visits varied based on the attending clinicians’ direction.
Data related to treatments conducted within the first 27 months of study startup were assessed. Safety data were collected for an additional 3 months, to allow for sufficient follow-up of subjects treated near the study end date.
Waiver of informed consent
As this research involved secondary use of information originally collected for charting purposes, it would have been impracticable to conduct this study without a waiver for informed consent. Therefore, the researchers requested a waiver of consent that was in accordance with the standards described by the Government of Canada and the Canadian Institute of Health Research. The waiver of consent was further justified because the research involved no risk to patients, the lack of the patient’s consent was unlikely to adversely affect the welfare of the patients (as they are treated as per routine care) and the study did not involve administering a therapeutic intervention, but rather evaluated information after patients had been routinely treated by their attending clinicians. Furthermore, this research satisfied all the criteria in Article 5.5 Parts A to F regarding researchers who use secondary data without informed consent. It also conformed to industry standards whereby no consent is obtained as this was a limited data set and does not include identifiable information.
Sample size and recruitment
Given the study design (ie, capturing all available cases in the population), no traditional sample size calculation was performed. However, based on previous reports regarding the incidence rates of rare AEs in patients following injectable treatments (eg, 0.004% for necrosis),1-14 it was expected that data relating to ~200 AEs would be entered.
It was estimated that approximately 10 Canadian sites would participate in the study. Recruitment of study sites was determined following the administration of a site feasibility questionnaire (Supplemental Material 4).
Procedures
Clinicians accessed the portal via an online platform, after successful completion of the site screening questionnaire. Each user was provided with a unique username and password.
Results
Study Centers
In total, 10 sites participated in the present study. These sites were location in Quebec (n = 3), Ontario (n = 3), New Brunswick (n = 1) and the West Coast [British Columbia, Alberta, Manitoba (n = 3)].
Adverse Events
Throughout the active phase of the trial, 123,124 injectable treatments were conducted. One hundred and eleven patients, experiencing a total of 235 AEs, were entered into the portal. This equated to an AE incidence rate of 0.19% (235/123,124), per treatment. Thirty unique products were associated with AEs, including 2 biostimulators, 3 neurotoxins, and 25 hyaluronic acid-based fillers. In total, there were 112/235 (47.66%) mild AEs, 88/235 (37.45%) moderate AEs, and 35/235 (14.90%) severe AEs. The most common complication (n = 48/235; 20.43%) was swelling, with a per treatment prevalence of 0.04%. Of the 235 documented AEs, only 5 (2.12%) were reported to other sources, including 1 case being reported to Health Canada and 4 cases to the respective product manufacturer.
Discussion
Innovative Solution for Improving Health Outcomes
One of the strongest breakthroughs in information and communication technology for the healthcare system was the introduction of electronic patient records. This is reflected in the increase of resources that the healthcare industry allocates to healthcare informatics from 2% of its revenues during the 1990s to 5% to 7% in more recent years. 23 Despite the fact that EDC systems have been available for over 2 decades, many clinical trials and practices are still mainly conducted collecting patient data using paper documents as the primary tool. In fact, it has been reported that in over 75% of practices, paper data collection is the main source of data acquisition. 24 A reason for this has been partially ascribed to the fact that standards have not been extended to facilitate data collection at the investigation site, as AE capture and reporting is one of the most time-consuming activities related to the conduct of trials and in clinical practice. Another reason relates to the fact that present technological applications often do not have adequate functionality to meet current needs. Therefore, there remains a high demand for an EDC system that is designed to be quick, easy to deploy, simple to use, captures true AEs, as defined in Edwards and Aronson 2000, as “an appreciably harmful or unpleasant reaction, resulting from an intervention related to the use of a medicinal product, which predicts hazard from future administration and warrants prevention or specific treatment, or alteration of the dosage regimen, or withdrawal of the product,”23,24 and ensures long-term capture of safety outcomes for injectable products. 25
Herein, we describe the development and validation of an electronic tool that can be used in clinic settings for real-time AE collection (Figure 1). The EDC software can offer several benefits including regulatory compliance, help organizations manage pharmacovigilance requirements, ensure data quality, integrity, and transparency, and improve patient safety. The EDC system may also lower the costs of collecting, storing, and distributing clinical trial data, as well as modernizes the data collection process. To date, the platform has been validated through a pan-Canadian observational study, with its initial deployment into 10 medical-aesthetic clinics. Of note, the usefulness of the registry was evidenced by the significant increase in the number of AE reports submitted by sites, in comparison to reports submitted by the same sites via other means. For example, of the 235 documented AEs, only 5 (2.12%) were reported to other sources, including 1 case being reported to Health Canada and 4 cases to the respective product manufacturer.
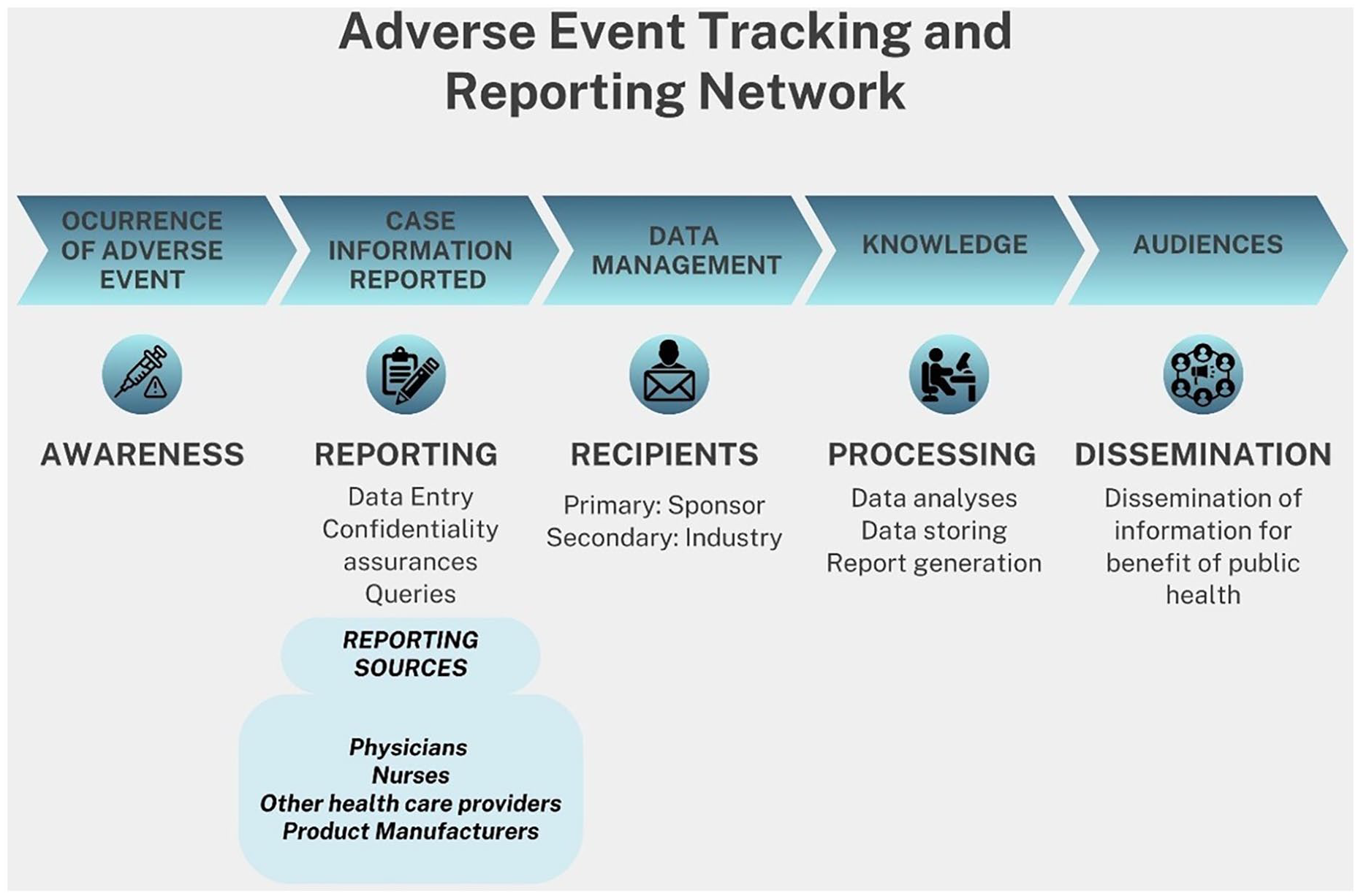
Flow chart of registry procedures.
Implications
The validation and implementation of the GRACE Portal offers many near and far-reaching benefits, including creating a cohort of subjects for future epidemiological, health services and outcomes research; enabling clinics to compare their performance with aggregated data; supporting clinicians in decision making and in quality improvement activities; helping decision-makers evaluate policies; and encouraging medical practice change and improved clinical outcomes.
Herein, the true incidence of any AE following an aesthetic injectable treatment was found to be 0.19%, which is higher than previous reports. This finding stresses the importance of re-evaluating current AE protocols, and validating treatment recommendations through clinical trials.
Future Directives
Future directives include increasing outreach strategies to implement the GRACE Portal throughout Canada and internationally, creating algorithms or protocols for the management and/or treatment of AEs and standardizing care, and ultimately, contribute to the improvement of healthcare outcomes in aesthetic patients. The possibility and benefit of making the portal tablet and phone compatible is being evaluated.
Supplemental Material
sj-docx-1-cms-10.1177_12034754241311270 – Supplemental material for Global Registry of Adverse Clinical Events (GRACE©): A Prospective, Multicenter, Observational Cohort Evaluating Complications Associated with Aesthetic Injectables
Supplemental material, sj-docx-1-cms-10.1177_12034754241311270 for Global Registry of Adverse Clinical Events (GRACE©): A Prospective, Multicenter, Observational Cohort Evaluating Complications Associated with Aesthetic Injectables by Kaitlyn M. Enright, Andreas Nikolis and John Sampalis in Journal of Cutaneous Medicine and Surgery
Footnotes
Acknowledgements
We would like to thank the following clinician-investigators for participating in the present study: Drs. Demetrios Rizis, Martin Braun, Lisa Kellett, Sean Rice, Jack Kolenda, Andrei Metelitsa, Zaki Taher, and Chantal Chiasson.
Author Contributions
Kaitlyn M. Enright confirms responsibility for study conception and design, literature review, statistical analyses, interpretation of findings, and drafting of the manuscript. All authors revised it critically for important intellectual content, approved the final version to be published, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Statements and Declarations
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by a doctoral research award from The Fonds de recherche du Québec - Santé (FRQS).
Ethical Considerations
This study received approval by a central research ethics board (REB; Institutional Review Board Services) and, where applicable, also local REB approval (HREBA). The IRBs determined that the Canadian privacy requirements for a waiver of consent were met. The study was carried out in accordance with Health Canada regulations, 21 CFR parts 56 and 312.3 and 45 CFR 46, good clinical practices (eg, ICH GCP Guidelines), Alberta Health Information Act, and the Tri-Council Policy Statement for Ethical Conduct of Research Involving Humans, as appropriate to the research. Copies of the REB certifications and a description of the research were sent to the Office of the Information and Privacy Commissioner (OIPC).
Consent to Participate
Not applicable.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.