
Abstract
Background:
The performance and genetic role in host response delineate investigative points of polymorphisms as potential biomarkers in viral infections.
Methods:
Thus, this research aimed to map biomarkers and risk factors in the severity of COVID-19 in individuals in Western Amazon (n = 243).
Results:
Patients aged 40 to 59 years showed an association with clinical progression (P = .003), also evidencing the relationship for individuals >60 years (P < .001), besides the non-vaccination influenced the pathology (P = .023). qPCR for human genotyping of the targets rs2070788, rs4702, rs76635825, rs540856718, rs35803318, rs12979860, and rs16899066, as well as for gene expression of ACE2, HLA-A, HLA-B, IFNL-3/2, IL-6, and TMPRSS2 was used. The rs12979860 (C > T) and rs2070788 (A > G) showed association among the analyzed groups (P < .05) with the allelic and genotypic frequency of rs12979860 (x2 < 3.84) and evolutionary pointing of rs2070788G allele among infected people, including deaths.
Conclusion:
Gene expression showed high levels between the moderate and severe groups, with emphasis on TMPRSS2 and IL-6 genes that performed better. Thus, there is possibly an association regarding the role of the TMPRSS2 gene and rs2070788G, as well as age and IL-6 levels for COVID-19, pointing in parallel to the considerable influence of the vaccine on the SARS-CoV-2 pathway.
Background
Severe Acute Respiratory Syndrome Virus 2 (SARS-CoV-2) has a high transmissibility profile, which rapidly led to a global pandemic, resulting in approximately 768 million infections and nearly 7 million deaths from COVID-19.1 -3 In Brazil, Coronavirus Disease 2019 (COVID-19), caused by SARS-CoV-2, has led to over 38 million reported cases and more than 700 000 deaths. 4 This disease can manifest in various forms, ranging from mild symptoms to the development of severe acute respiratory syndrome (SARS), 5 however, several factors can influence the clinical progression of the disease, including age, comorbidities, and host genetics.6,7
Many human genes associated with viral infections are linked to single nucleotide polymorphisms (SNPs).8 -11 SNPs, characterized as point variations, are among the most common types of genetic variation in the human genome.6,12 These variations can influence human responses to infections by altering gene expression, function, and the affinity of endogenous structures through different mechanisms, thereby contributing to susceptibility or resistance to various diseases, including COVID-19.13 -15
Genome-wide association studies (GWAS) have identified several genes linked to increased susceptibility to viral infections, like COVID-19. 16 Polymorphisms are most frequently associated with ACE2, a viral tropism protein,17 -19 TMPRSS2 and FURIN, proteases involved in the cleavage and activation of the spike protein.20,21 Other studies highlight genetic influences on immune response, from antigen recognition and presentation via the Human Leukocyte Antigen (HLA) complex, which initiates an effective immune response, 22 to the severity of infection, driven by cytokine storm proteins like Interleukin-6 (IL-6) and the antiviral response mediated by IFNL3, which has been linked to the severity of respiratory infections caused by other viruses.23 -25
This study conducted a molecular genetic analysis of polymorphisms in the ACE2, TMPRSS2, FURIN, HLA, IL6, and IFNL3 genes to evaluate genotypic distribution and gene expression patterns in the population of Rondônia, in the Western Brazilian Amazon. The aim was to understand the influence of potential biomarkers on the clinical progression of COVID-19.
Methods
Ethical aspects
The research was conducted under protocol number 4 000 086 and CAAE: 30915320.0.0000.0011, approved by the Research Ethics Committee of the Centro de Pesquisa em Medicina Tropical de Rondônia (CEPEM/RO). Written informed consent was obtained from all participants and/or their legal guardians prior to sample collection, and all experiments were performed in accordance with relevant guidelines and regulations.
Study population
The present work is a descriptive and cross-sectional analysis conducted from March to August 2022 in collaboration with the reference hospital for SARS-CoV-2 cases in Rondônia, the Centro de Medicina Tropical de Rondônia (CEMETRON). All procedures were performed in the Molecular Virology Laboratory and the Laboratory of Cellular Immunology Applied to Health at the Oswaldo Cruz Foundation, Rondônia (FIOCRUZ/RO). We evaluated 223 individuals of both sexes, all of whom had a molecular diagnosis of SARS-CoV-2 confirmed by RT-qPCR (performed by the Central Public Health Laboratory of Rondônia, LACEN/RO) and/or antigen testing (COVID-19 Ag Rapid Test Kit—Instituto de Biologia Molecular do Paraná), Pregnant women, Indigenous people, individuals with HIV, and those with a negative diagnosis for COVID-19 were excluded from the study. The participants, who were positive for SARS-CoV-2 and potentially diagnosed with severe acute respiratory syndrome (SARS), were stratified into 3 clinical categories according to established protocols 26 : mild, moderate, and severe. A control group of 20 samples, stored in a biorepository, consisted of individuals who had never been exposed to SARS-CoV-2.
The socio-demographic profile of moderate and severe cases was analyzed using data collected through CEMETRON’s hospital-integrated system (HOSPUB). Data collection related to the characterization of severe acute respiratory syndrome (SARS) among hospitalized individuals followed the established clinical management guidelines for hospitalized cases. 27 For mild cases, data were collected using the Laboratory Environment Manager (LEMM) and through direct consultation by completing an epidemiological investigation form. Vaccination status was classified as fully vaccinated (complete vaccination schedule with or without booster doses), partially vaccinated (incomplete vaccination schedule), and unvaccinated.
Evaluation of the genetic profile
Genomic deoxyribonucleic acid (gDNA) was extracted from whole blood samples using a commercial PureLink® Genomic DNA Mini Kit (Thermo Fisher Scientific). All samples were analyzed by qPCR following kit optimization and were used for genetic profiling. The reaction was performed with 1X TaqMan® Universal PCR Master Mix (2X) and 1X TaqMan® Genotyping Assay Mix (40X) (Thermo Fisher Scientific, Massachusetts, USA) for the SNP assays listed in Table 1, along with 30 to 40 ng/µl of extracted DNA, resulting in a final volume of 25 µl. The cycling conditions for the reaction were as follows: pre-reading at 60°C for 30 seconds, activation of DNA polymerase at 95°C for 10 minutes, followed by 45 cycles of denaturation at 95°C for 15 seconds, and annealing/extension at 60°C for 60 seconds, concluding with a post-reading at 60°C for 30 seconds.
Identification of assays for polymorphisms. List of targets used during the human genotyping process, indicating the sequence for each marker and the genes where the SNPs are located.

For data analysis, we used the genotyping module of the Design & Analysis Software v2.6.0 (Thermo Fisher Scientific) with a 95% confidence interval, automated threshold, and Baseline from 5 to 15.
Gene expression for all patients participating in the project
The blood samples underwent extraction to obtain tRNA using the commercial SV Total RNA Isolation System (Promega, Madison, WI, USA), following the manufacturer’s instructions. To measure gene expression via RT-qPCR, custom TaqMan® Gene Expression Assay (20X) kits were utilized for the following assays: Hs04193048_gH (IFNL3/2), Hs00174131_m1 (IL-6), Hs05024838_m1 (TMPRSS2), Hs07292706_g1 (HLA-B), Hs01058806_g1 (HLA-A), and Hs01085333_m1 (ACE2). The reaction was performed using 1X TaqMan® Fast Virus 1-step Master Mix (4X) (Thermo Fisher Scientific, Massachusetts, USA) combined with 1X of the target assay and 5 to 10 ng/µl of extracted RNA, resulting in a final volume of 10 µl. Normalization of the endogenous gene was carried out using the geNorm algorithm and the ExpressionSuite software (version 1.3) with 2 candidate genes: 4333769F (18S ribosomal RNA) and 4333760F (TATA box). The cycling conditions were as follows: reverse transcription at 50°C for 5 minutes, activation of DNA polymerase at 95°C for 20 seconds, followed by 40 cycles of denaturation at 95°C for 15 seconds and annealing/extension at 60°C for 1 minute. Data analysis was conducted using the Relative Quantification module of the Design & Analysis Software (Version 2.6.0, Thermo Fisher Scientific), with a confidence interval of 95%, a threshold of 0.3, and a baseline set from 3 to 15 for all targets and assays. The 2−ΔΔCt method was applied to calculate fold changes, with the housekeeping gene 18S used for normalization.
Statistical analysis
Data were analyzed using R software (v4.0.3) and GraphPad Prism v10.1.2. Fisher’s Exact Test was applied, with P-values <.05 considered statistically significant. To assess allelic and genotypic frequencies of the polymorphisms, the chi-square test (X²) was performed to evaluate Hardy-Weinberg equilibrium, with a significance level set at 3.84 and 1 degree of freedom.
Results
The study sample consisted of 223 participants, classified as follows: 50 with severe, 150 with moderate, and 23 with mild clinical status. Of these, 200 individuals were from the Centro de Medicina Tropical de Rondônia (CEMETRON/RO), a reference hospital for moderate and severe COVID-19 cases, while 23 were identified as mild cases from Primary Health Care (PHC) units in Porto Velho, RO.
The prevalence of male participants was 61.88% (138/223), though no significant difference was observed (P = .149), as shown in Table 2. However, statistically significant differences were found between the age groups 40 to 59 years, those over 60, and vaccination status.
Frequency of variables according to clinical progression to COVID-19.

Statistical significance in Fisher’s exact test.
An association analysis of genetic variables among the 7 polymorphisms across the groups yielded 1701 results. Among the markers, rs2070788 showed a predominance of the GG genotype, while the CT heterozygous genotype was most prevalent for rs12979860. Both polymorphisms demonstrated significant associations between genotype frequency and the groups (P = .0015 and P = .0013, respectively), as shown in Figure 1.

The genotype distribution between polymorphisms and the clinical groups was evaluated, revealing variability among the analyzed SNPs, except for rs540856718 in the Human Leukocyte Antigen (HLA), which showed no allelic variation. Statistical significance was observed in Fisher’s Exact Test for rs12979860 and rs2070788.
The Hardy-Weinberg test revealed that rs12979860 was in equilibrium across all clinical groups (X² < 3.84). However, rs2070788 showed disequilibrium among the mild, moderate, and severe groups, with a complete absence of heterozygotes in the control group (Table 3).
Hardy Weinberg equilibrium distributed among the polymorphisms.

OHbs corresponds to the observed heterozygosity.
Reference allele
Mutant allele
In the severe group, 46% (23/50) of the patients died. Most of the deceased were male (65.21%), with 56.53% (13/23) identifying as brown, and the average age was 62 years. The average hospitalization time was approximately 12 days, ranging from 2 to 31 days. Among this group, 16 individuals were fully vaccinated, with an average hospital stay of 11.06 days, which was shorter compared to non-vaccinated individuals, who had an average stay of 14.8 days.
A total of 65.21% (15/23) of the patients had comorbidities, with some presenting more than one condition. The most common were diabetes and hypertension, followed by severe chronic lung diseases, neurological disorders, immunosuppression, and cardiovascular diseases.
Regarding the genetic profile within the group, a high proportion of the GA genotype of rs73635825 was observed in both male and female individuals (Figure 2), as well as the CT genotype of SNP rs12979860. These findings were similarly distributed across vaccination status. The presence of the rs2070788G allele was significant among the deceased patients. Although genotype distribution was assessed, no statistical significance was found (P = .4628).

Distribution of genotypes and immunization profiles among polymorphisms in the group of individuals who died. Genotypes were schematized by different symbols and immunization groups were differentiated by colors.
Gene expression analysis was conducted on 29.62% (72/243) of the samples, distributed across groups with varying degrees of symptomatology for 6 different targets, in addition to the control group and the stratification of deaths. Low ACE2 gene expression was consistently observed in all evaluated groups (Figure 3), with significant expression found in only 6.94% of the cohort. HLA-A levels remained stable across all analyzed groups. IL-6 and HLA-B exhibited similar detection levels between the moderate and severe groups; however, IL-6 levels significantly equilibrated among the mild, control, and deceased groups.
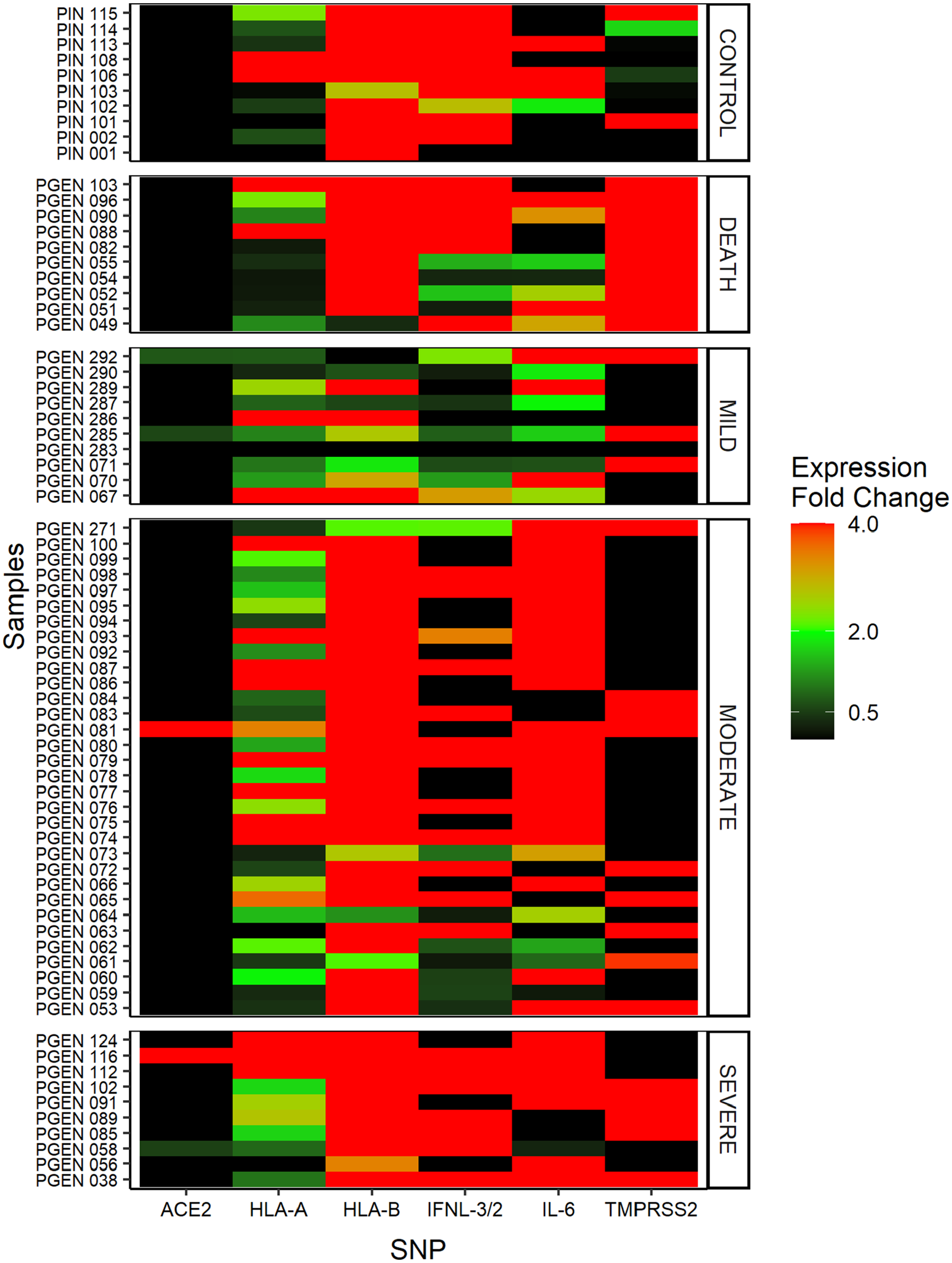
RT-qPCR for gene expression and comparative graph of target gene levels and the clinical progression of COVID-19. Graph of the relationship of gene markers with the clinical progression of COVID-19. Gene expression values were obtained by calculating 2−ΔΔCt. Overexpression of samples was considered at fold change values above 2. The representations were demonstrated by color intensity. Created with Biorender.com.
The performance of TMPRSS2 indicated that this target is actively expressed within the population, with normal expression detected in only one individual from the control group. Notably, in the cases of deceased patients, overexpression of this gene was observed in all individuals.
Regarding the frequencies of the analyzed genotypes, rs2070788 displayed a difference only in comparison to the direct analysis of TMPRSS2 gene expression. Among all groups, only the mild group differed concerning the persistence of low expression levels detected in individuals genotyped as GG, as illustrated in the analyses below (Figure 4). The control group showed a balanced comparison of genotypes and expression levels. Additionally, the polymorphisms and their genetic variations with higher frequencies in the general population did not diverge when related to increased fold change values.

Comparison between SNP genotypes and gene expression. Direct association between the expressed levels of all genes analyzed, compared to the distribution of genotypes for each polymorphism.
Discussion
In this study, human polymorphism genotyping and gene expression tests were conducted to assess associations with the progression profile of COVID-19 in individuals from a state along the Brazil-Bolivia border. The TMPRSS2 gene was found to play a significant role in case progression, while rs12979860 and rs2070788 indicated a direct association with the clinical groups. Notably, the rs2070788G substitution allele may have a predominant influence in the population.
The state of Rondônia has historically been affected by migratory movements from interstate regions within Brazilian territory, as well as by populations from neighboring countries such as Bolivia and more distant countries like the United States of America (USA). 28 The geographic characterization of the state as a border region and the construction of the Madeira-Mamoré railway, aimed at overcoming the difficulties of river traffic on the Madeira and Mamoré rivers and facilitating the transportation of rubber and other products from the Amazon region, were crucial for the establishment of the current highly mixed population.29,30
Genome-Wide Association Studies (GWAS) report the association of genetic variations, such as single nucleotide polymorphisms (SNPs), one of the most common forms of polymorphism, with susceptibility to infection and the pathological clinical progression of COVID-19 to more severe cases. 31 Genetic variations in genes responsible for modulating the immune response are among the main targets of immunogenetic studies of this disease, where SNPs such as rs12979860 and rs368234815 in the Interferon Lambda 3 and 4 (IFNL3 and IFNL4) genes are of particular interest 32 and rs1800795 in the Interleukin-6 (IL-6) gene, 33 have been associated with the severity of COVID-19 in certain populations.
It has been reported that SARS-CoV-2 infections occur in individuals who have completed their vaccination doses, 34 this characteristic was observed in this study. However, among the moderate group evaluated, the highest percentage of fully vaccinated individuals was noted compared to the other groups. This finding aligns with other studies regarding the effectiveness of booster doses in reducing the severity of the disease.33,35,36 When evaluating hospitalization duration among patients, the group that did not receive any doses of the vaccine had a longer average stay of 14.8 days. This finding supports studies that highlight the protective effect of vaccination, even for individuals hospitalized with COVID-19. 37
The predominance of males in this study differed from findings in other studies conducted in the same region of Brazil. In contrast to the genomic investigation results, the cases were categorized into clinical outcome groups. This sex-related characteristic may be influenced by the potential direct action of germline genes,38,39 the mutational variety occurring in genes located on sex chromosomes can directly contribute to advancing the clinical understanding of diseases. 40
For the genes studied, certain polymorphisms were anticipated in a comparative population. For instance, SNP rs540856718 of HLA-DPA1 showed no allelic diversification among all evaluated individuals, directly impacting the analysis of equilibrium due to the absence of representatives. This type of profile is not considered atypical from a population genetics perspective 41 fixed statistics of allelic and genotypic frequencies can indicate that a given group is not experiencing pressure from the external environment.
Like the HLA-DPA1 polymorphism, the same finding was observed for rs35803318 of ACE2, which exhibited only the CC genotype in all individuals within the mild population. Generally, in such cases, genetics suggests that those affected are in equilibrium regarding factors that directly influence disease progression. Notably, this SNP was the only one associated with infection in an Iranian population. 42 In a study evaluating ACE2 variants through exome analysis in the Madrid population, the authors found that this SNP typically does not exhibit homozygosity among individuals infected with SARS-CoV-2. 43 This finding differs significantly from observations in the Brazilian population, despite the use of different methodologies.
The Hardy-Weinberg equilibrium test indicated that rs12979860 was stable across all categorized outcomes. This characteristic supports the investigation of adverse situations reflected in other results. Similar to the adaptations observed among isolated populations, such as the Bajaus or “Sea Nomads,” who possess an essential ability to dive, this stability may indicate a genetic adaptation to environmental challenges, 44 this population genetic balance can be hypothesized, as it was also found to be statistically significant. The statistics for rs12979860 of IL-28b were predictive, while homozygosity of the substitution allele was identified as a characteristic of rs2070788 in the TMPRSS2 gene. This finding suggests that the G substitution allele is dominant in the population, overshadowing the association of the A allele with this polymorphism. Among all vaccination groups, the GG genotype was the most prevalent, with 48.92% of individuals representing the highest level of dispersion among those studied, specifically among those who were fully vaccinated. This genotype was found in 33.33% of partially vaccinated individuals, followed by 39.6% of non-vaccinated individuals. Furthermore, heterozygotes were also well represented, comprising 20.14% of the fully vaccinated group, 22.22% of the partially vaccinated group, and 30.2% of the non-vaccinated group. The expected AA genotype showed low prevalence compared to the predominance of the G allele across all groups. Among the fully immunized, the AA genotype represented 30.94%; among the partially vaccinated, it was 44.45%; and among the non-immunized, it accounted for 30.20%.
Although rs2070788 has been previously studied in a population in Southern Europe, 45 and showed no association with prolonged COVID-19 symptomatology, one study noted that the G allele of this polymorphism is linked to the ancestral Native American population, 46 this characteristic indicates a non-mixing population. In contrast, while the association between this variation of the TMPRSS2 gene and COVID-19 was assessed in a non-mixed population, no statistical association was found. However, in this research, a different data profile emerged, revealing statistically significant values and tabulated evolutionary parameters. Additionally, the expression of this gene showed overexpression, particularly among the deceased cases, across the 4 clinical groups evaluated.
In the case of the elevated expression of TMPRSS2 observed in the severe group, particularly among the deceased, the potential role of TMPRSS2 in COVID-19 may be linked to the rs2070788 and rs383510T polymorphisms, which are associated with increased protease expression in human lung tissue.6,47 When associated with the G allele of rs2070788, the expression levels of TMPRSS2 remained elevated, a characteristic observed in most individuals. To explain this phenomenon, studies indicate a higher risk for males linked not only to polymorphisms but also to androgen disposition. This observation is further supported by the association between hair loss and male individuals with COVID-19 who were hospitalized.6,48 -50 Interestingly, the TMPRSS2 promoter gene contains a 15 bp androgen receptor (AR) response element, suggesting that the human protease is regulated by these receptors. This regulation indicates potential gender differences in SARS-CoV-2 infection.6,51
In the hospitalized groups, the elevated levels of IL-6 gene expression observed align with findings from Brazilian researchers who conducted a multivariate study on IL-6 expressions, 52 the estimated elevated values identified in this research may be associated with the regulation of non-coding RNAs (ncRNAs) and the IL-6 signaling cascade triggered by this action.
Conclusion
This study demonstrated that vaccination was crucial in preventing the aggravation of infection in these individuals. The stability of certain genes indicates a genetic balance within a population that may be undergoing continuous adaptive and/or evolutionary processes over the years. Furthermore, the expression levels of the TMPRSS2 gene among the stratified groups raise questions about classifying this gene as a potential marker for the evolutionary dynamics occurring in the interaction between individuals and SARS-CoV-2. Notably, there appears to be a significant relationship between the rs2070788G allele and the mapped individuals, prompting discussions regarding the adaptation process in response to external environmental pressures. Our study had a primary limitation related to the sample size, however, corresponding to the demand for hospital admissions at the reference hospital for treating COVID-19 cases in the state of Rondônia during the study period; however, we provided information on the genetic profile of the population of western Brazilian Amazon, as well as the clinical characteristics associated with genetic aspects in COVID-19, offering data on the features of this highly mixed region.
Supplemental Material
sj-xlsx-1-bec-10.1177_11795972241298786 – Supplemental material for Screening Biomarkers and Risk Factors for COVID-19 Progression in a Border Population Between Brazil-Bolivia
Supplemental material, sj-xlsx-1-bec-10.1177_11795972241298786 for Screening Biomarkers and Risk Factors for COVID-19 Progression in a Border Population Between Brazil-Bolivia by Ana Maísa Passos-Silva, Adrhyan Araújo, Tárcio Peixoto Roca, Jackson Alves da Silva Queiroz, Gabriella Sgorlon, Rita de Cássia Pontello Rampazzo, Juan Miguel Villalobos Salcedo, Juliana Pavan Zuliani and Deusilene Vieira in Biomedical Engineering and Computational Biology
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Fundação Oswaldo Cruz de Rondônia – FIOCRUZ/RO (Process: VPGDI-008-FIO-21-2-22-17 PROGRAMA DE EXCELÊNCIA EM PESQUISA DA FIOCRUZ RONDÔNIA – PROEP), and by Instituto Nacional de Epidemiologia da Amazônia Ocidental - INCT EpiAmO.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Conceptualization: AMPS, DV; Data processing and analysis: AMPS, AA; Investigation: AMPS; Methodology: AMPS, AA, JASQ; Project Administration: DV; Supervision: DV, JPZ; Writing–original draft: AMPS, AA, TPR, JASQ, GS; Writing–review and editing: AMPS, DV, JMVS, JPZ, RCPR. All authors reviewed the manuscript.
Data Availability
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.