
Abstract
Human immunodeficiency virus (HIV) is an infectious virus that depletes the CD4+ T lymphocytes of the immune system and causes a chronic life-treating disease—acquired immunodeficiency syndrome (AIDS). The HIV genome encodes different structural and accessory proteins involved in viral entry and life cycle. Determining the 3D structure of HIV proteins is essential for new target position finding, structure-based drug designing, and future planning for computational and laboratory experimentations. Hence, the study aims to predict the 3D structures of all the HIV structural and accessory proteins using computational homology modeling to understand better the structural basis of HIV proteins interacting with host cells and viral replication. The sequences of HIV capsid, matrix, nucleocapsid, p6, reverse transcriptase, invertase, protease, gp120, gp41, virus protein r, viral infectivity factor, virus protein unique, RNA splicing regulator, transactivator protein, negative regulating factor, and virus protein x proteins were retrieved from UniProt. The primary and secondary structures of HIV proteins were predicted by Expasy ProtParam and SOPMA web servers. For the homology modeling, the MODELLER predicted the 3D structures of HIV proteins using templates. Then, the modeled structures were validated by the Ramachandran plot, local and global quality estimation scores, QMEAN scores, and Z-scores. Most of the amino acid residues of HIV proteins were present in the most favored and generously allowed regions in the Ramachandran plots. The local and global quality scores and Z-scores of the HIV proteins confirmed the good quality of modeled structures. The 3D modeled structures of HIV proteins might help further investigate the possible treatment.
Keywords
Introduction
Human immunodeficiency virus (HIV) is an infectious virus that depletes the CD4+ T lymphocytes of the host immune system, which leads to a chronic life-treating disease- acquired immunodeficiency syndrome (AIDS). 1 According to UNAIDS, in 2021, approximately 38 million people were living with HIV, and 650 000 people died from AIDS-related illnesses. 2 CD4+ receptors of helper T cells such as macrophages, eosinophils, and neutrophils facilitate the HIV binding and entry to the cells. 3 The genome of HIV has 9 genes that encode 15 viral proteins. The polyprotein of HIV includes Gag proteins (capsid, matrix, nucleocapsid, and p6), Pol proteins (reverse transcriptase, invertase, protease), Envelope glycoproteins (Gp120, Gp41), and accessory proteins (virus protein r, viral infectivity factor, virus protein unique, RNA splicing regulator, transactivator protein, negative regulating factor, and virus protein x proteins) cleaved by cellular proteases. 4 When the HIV-1 virus enters a cell, the envelope glycoproteins facilitate the fusion of the virus and cellular membranes. 5
In HIV, capsid protein (p24) forms the conical capsid, matrix protein (p17) forms the inner membrane layer, nucleocapsid (p7) intricates in the formation of RNA complex, and p6 involves in the release of virus particles. 6 Protease (p10) involves the proteolytic cleavage of precursor protein resulting in structural proteins and viral enzymes. Reverse transcriptase (p51) transcribes the RNA of HIV into DNA. Integrase (p32) protein integrates the proviral DNA into the host genome. Tat (p14) activates the viral gene transcription. Rev (p19) regulates the export of non-spliced and partially spliced viral mRNA. 7 Nef (p27) influences HIV replication, enhancement of infectivity of viral particles, and downregulation of CD4+ on target cells. Vif (p23) is critical for infectious virus production. 7 Vpr (p15) interacts with p6, facilitates virus infectivity, and affects the cell cycle. Vpu (p16) controls CD4+ degradation and modulates intracellular trafficking. Vpx (p15) is involved in the early steps of virus replication of HIV-2. 8 Gp120 (surface glycoprotein) facilitates the attachment of the virus to the target cell. In vitro studies confirmed that the recombinant Gp120 interacts directly with CD4+/co-receptors and activates the cellular pathways involving actin regulation, proliferation, cell adhesion, and increased chemokines and cytokines. 9 Gp41 (transmembrane protein) intricates the anchorage of gp120, the fusion of viral and cell membranes. Gp120 interferes with CD4+ costimulatory functions and induces apoptosis in cells. 10 HIV-1 enters the target host cells through gp120 and gp41 trimeric complexes of viral envelope glycoproteins. 11 When gp120 binds to the CD4+ glycoprotein on target cells, it undergoes conformational changes that allow it to bind to either the CCR5 or CXCR4 chemokine receptors. 12 Gp41 plays a significant role in driving the fusion process by forming a fusogenic 6-helix bundle structure. 13 It has an ectodomain, a membrane-spanning segment, and a cytoplasmic tail. 14 After receptor binding, the transmembrane envelope glycoprotein gp41 exposes its ectodomain, causing additional conformational changes that result in the fusion of the viral membrane and host cell membrane. 12
The structural basis of HIV envelope protein interacting with host cells must be understood to identify new drugs that prevent HIV entry. 15 The prediction of reliable 3D protein structures has been successfully achieved by computational methods, allowing scientists to understand the protein’s behavior and function, its interactions with its ligands, and the effects of specific insertions, mutations, and deletions on its conformation and function.16,17 In previous studies, the simulated structures were collected by NMR spectroscopy and X-ray to reveal common structural motifs in V3 loops of HIV Gp120 protein. 18 By comparative modeling and simulated annealing, the amino acid sequence of the V3 loop and its conformation in 3D folds and local geometry has been determined. 18
The structural characteristics of proteins play a crucial role in elucidating the molecular mechanisms underlying biological processes. 19 Crystallography and Nuclear Magnetic Resonance (NMR) spectroscopy are generally used to develop quality protein structures, but these processes are time-consuming, expensive, and difficult to perform for membrane proteins. 20 An alternative technique to develop protein structures is in-silico 3D structure prediction using homology modeling. 20 Homology modeling predicts the 3D structures of the target proteins by using the similarity between the target and template sequences. This computational technique is efficiently helpful in predicting membrane proteins such as HIV gp120 and gp41 that are difficult to crystallize. Further, homology modeling provides a clear understanding to study the receptor-ligand interaction. Single or multiple template modeling offers profound insight into unsolved structures but clinically significant drug-targeting proteins. As a result of homology modeling studies, various problems associated with crystallizing proteins that play essential roles in major disease pathways are often overcome and open the door to in-silico models providing more structural insight when experimental structures are lacking.21,22 Different studies have reported 3D-modeled structures of different proteins.23-26
Nevertheless, to our knowledge, no study has previously studied the 3D-modeled structures of all the HIV structural and accessory proteins. Yet, homology modeling is done by different online and offline servers and software. For instance, MODELLER is well known for its comparative tertiary structure prediction capabilities. 27 PROCHECK, Quantitative Model Energy Analysis (QMEAN), and Protein Structure Analysis (ProSA) servers are standard web servers for validating the modeled protein 3D structures.28,29 This study aims to use a computational method to predict the 3D structures of all HIV structural and accessory proteins to gain a better understanding of how HIV proteins interact with host cells and replicate. Further, different online tools validated the 3D modeled structures of HIV proteins to build other plots and graphs.
Methods
Proteins sequences
The RNA-based HIV-1 genome comprises all the information of 16 proteins required for the replication and structural assembly of the new virions. For example, the gag gene of the HIV genome transcribes 4 structural proteins, that is, capsid, matrix, nucleocapsid, and p6. In comparison, the pol gene transcribes proteins such as reverse transcriptase, invertase, and protease necessary for the replication of HIV. Further, surface glycoproteins gp120 and gp41 are transcribed from the env gene. All the structural and accessory proteins for HIV were listed, and their sequences were downloaded from UniProt (https://www.uniprot.org/) in FASTA format.
Primary structure prediction
The primary structures of all the HIV structural and accessory proteins were predicted in the Expasy ProtParam web server (https://web.expasy.org/protparam/). 30 In the primary structure analysis, different physiochemical characteristics such as aliphatic index (AI), base pairs length, extinction coefficient (Ec), grand average of hydropathy (GRAVY), instability index (II), molecular weight, theoretical isoelectric point (pI), and the total number of positive (+R) and negative (-R) residues were calculated.
Secondary structure prediction
The secondary structures of the HIV structural and accessory proteins were predicted in an online server SOPMA (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html). All the parameters of SOPMA were set by default, such as the numbers of conformational states to 4 (helix, sheet, turn, and coil), similarity threshold to 8, and window width to 17. 31
Homology modeling
HHpred online server (https://toolkit.tuebingen.mpg.de/tools/hhpred) was used for the homology modeling of all the targeted HIV proteins. HHpred used MODELLER after PIR alignment of the targeted sequence with the template(s) having maximum sequence similarity and identity.27,32 The MODELLER predicted the structure of the query sequence of the protein(s) with homologous protein(s) by using the far more conserved protein structure approach. Further, advanced structure modeling and loop refinement were performed to establish a 3D model of the protein structures. All the modeled structures were visualized in Discovery Studio Visualizer 2021.
3D structure validation of modeled proteins
All the modeled structures of HIV proteins were validated using methods such as the Ramachandran plot, local and global quality estimation scores, QMEAN scores, and Z-scores. PROCHECK online server (https://www.ebi.ac.uk/thornton-srv/software/PROCHECK/) was used to plot the Ramachandran plot for the conformational, stereochemical, and structural quality validation of the 3D modeled HIV structural and accessory proteins. 33 In the Ramachandran plot, about 90% of the amino acid residues in the most favored regions confirm the good quality of the 3D modeled structures. Similarly, for the estimation of both global (ie, for the entire structure) and local (ie, per residue) errors, the QMEAN server (https://swissmodel.expasy.org/qmean/) was used to calculate the composite scoring function of 3D modeled structures of HIV proteins. 34 The overall quality of an entire model is calculated by a global score. 35 Based on the alignment and template used in constructing the model, the Global Model Quality Estimation can be expressed as a number between 0 and 1, which reflects the expected model accuracy. The denominator of this number is the coverage of the target sequence. Reliability increases with higher numbers.36-38 For the local quality estimation of each residue, the model structure on the x-axis compares the expected similarity to the native structures on the y-axis. Residues that show a local quality score above 0.6 are considered good. 20 The QMEAN score above −4.0 confirms the good quality of the predicted 3D structures. Further, the ProSA tool (https://prosa.services.came.sbg.ac.at/prosa.php) evaluated the overall quality of each modeled structure of the proteins with a Z-score. The Z-score compared the quality of the 3D modeled proteins with all the experimental NMR and X-ray crystallographic protein structures. 28 The QMEAN Z-score estimates the degree of nativeness, such as inter-atomic packing, back-bone geometry, and unexpected solvent accessibility of the modeled structure with the quality of experimental structures for native proteins of similar size.28,29
Results
Protein sequences
The sequences of all the HIV structural (capsid, matrix, nucleocapsid, p6, gp120, gp41, reverse transcriptase, invertase, and protease) and accessory (virus protein r, viral infectivity factor, virus protein unique, RNA splicing regulator, transactivator protein, negative regulating factor, and virus protein x) proteins were retrieved from the UniProt database. Table 1 represents all the genes, proteins, their UniProtKB ID, and amino acid sequences of all the structural and accessory proteins of HIV.
HIV structural and accessory proteins with amino acid sequence and UniprotKB ID.
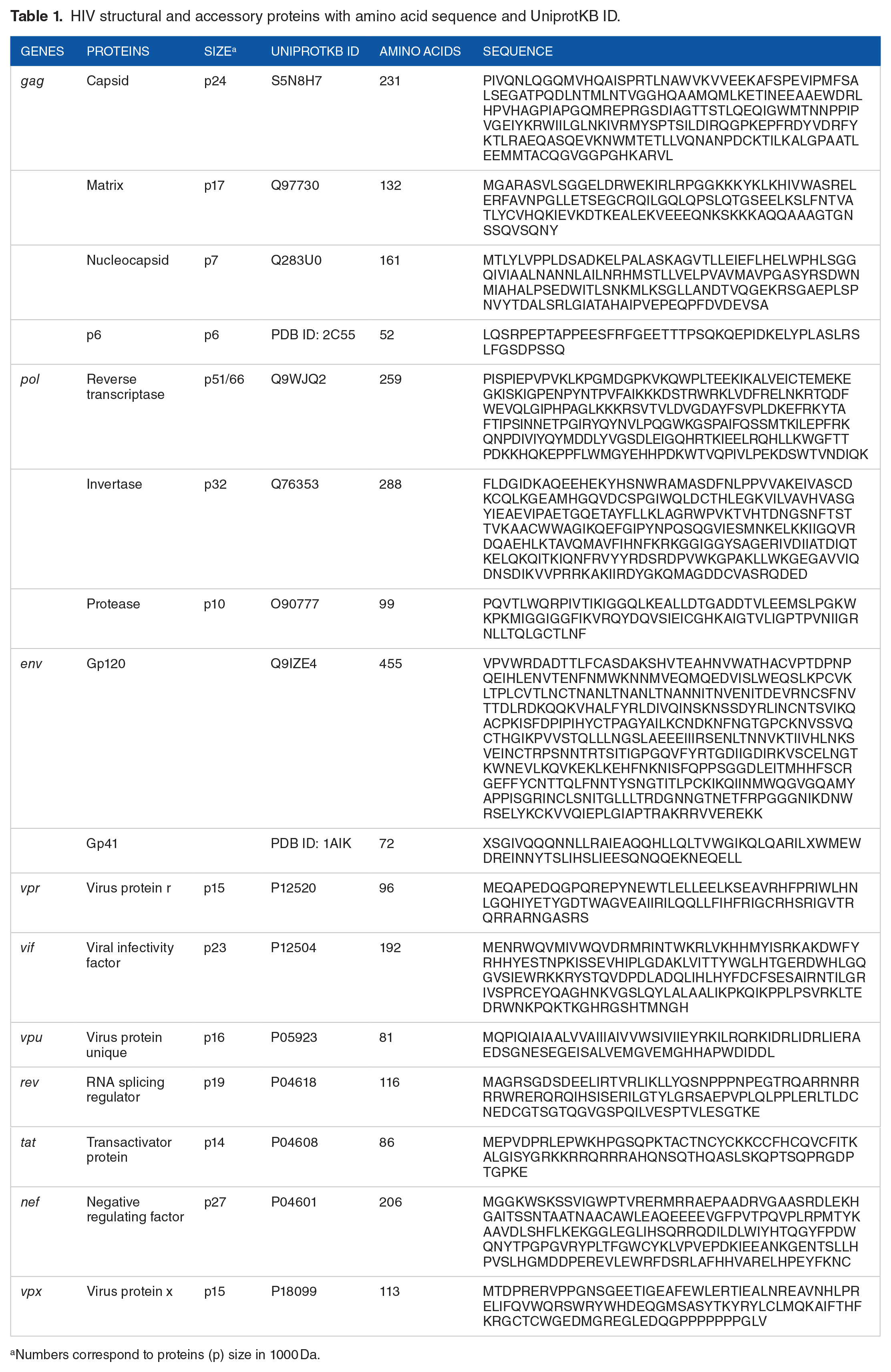
Numbers correspond to proteins (p) size in 1000 Da.
Primary structures of proteins
The ProtParam tool of the Expasy server predicted different physicochemical characteristics of HIV structural and accessory proteins. Table 2 represents the results of different physicochemical parameters of HIV structural and accessory proteins.
Physiochemical characteristics of HIV structural and accessory proteins.

Secondary structure prediction
Table 3 represents different values of the predicted parameters necessary for the secondary structures of HIV structural and accessory proteins using the SOPMA online server. All the secondary structures of HIV proteins showed alpha helix regions (except transactivator protein), extended strands, beta turns, and random coil. However, no HIV proteins confirmed the presence of 310 helices, pi helix, beta bridge, bend region, ambiguous states, and other states regions in their secondary structures.
Prediction of different parameters of secondary structures of HIV gp120 and gp41 proteins.

Homology modeling
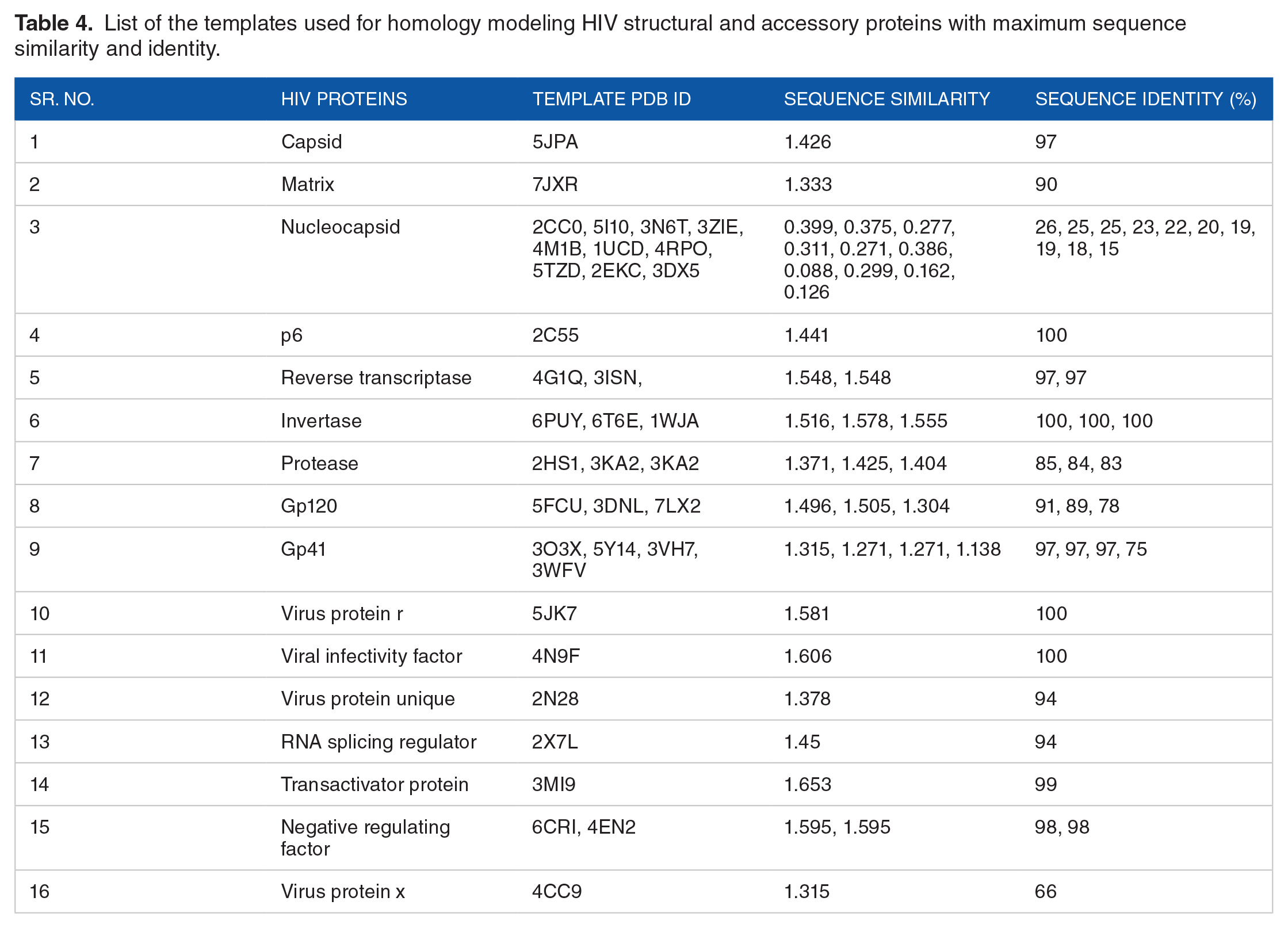
The templates used for the structure prediction of HIV proteins have maximum sequence similarity and identity obtained after using the BLAST program on Protein Data Bank. Table 4 shows the PDB IDs of the targeted templates of the respective HIV proteins with maximum sequence similarity and identity. For some proteins like HIV nucleocapsid, more than one template was used due to fewer sequence identities with the query sequences. MODELLER software predicted the 3D structures of all the HIV structural and accessory proteins. Figure 1 shows the 3D modeled structures of HIV structural and accessory proteins.
List of the templates used for homology modeling HIV structural and accessory proteins with maximum sequence similarity and identity.

Modeled 3D Structure of HIV Proteins: (A) capsid (p24), (B) matrix (p17), (C) nucleocapsid (p7), (D) p6, (E) reverse transcriptase (p51/66), (F) invertase (p32), (G) protease (p10), (H) GP120 ()) GP41, (J) virus protein R (p15), (K) viral infectivity factor (p23), (L) virus protein unique (p16), (M) RNA splicing regulator (p19), (N) transactivator protein (p14), (O) negative regulating factor (p27), and (P) virus protein x (p15).
3D structure validation of modeled proteins
The 3D modeled structures of HI structural and accessory proteins were validated by Ramachandran plot, local and global quality estimation scores, QMEAN scores, and Z-scores. Figure 2 shows the Ramachandran plot 3D modeled structures of HIV structural and accessory proteins. In the Ramachandran plot, the distribution of torsion angles of all the HIV structural and accessory proteins was analyzed to be present in favorable regions of the graphs. Table 5 displays the parameters of the Ramachandran plot, such as residues in the most favored region, residues additionally allowed region, residues generously allowed region, residues in the disallowed region, the total number of non-glycine and non-proline residues, numbers of end residues (excluding glycine and proline), numbers of glycine residues, numbers of proline residues, and the total number of residues in HIV structural and accessory proteins. QMEAN online web server calculated the local, global and normalized QMEAN4 scores of all the HIV 3D modeled protein structures. Figure 3 presents the local quality estimation graphs of HIV structural and accessory proteins. Figure 4 exhibits the global quality estimation scores of all the 3D-modeled HIV structural and accessory proteins. Figure 5 indicates the normalization of QMEAN4 scores of the 3D modeled structures of HIV structural and accessory proteins. The QMEAN scores below −4.0 are considered a low-quality of predicted structure. Figure 6 shows the plots of Z-scores of all the 3D modeled structures of HIV proteins by comparing the NMR and X-ray crystallographic structures. The Z-scores of HIV capsid, matrix, nucleocapsid, p6, reverse transcriptase, invertase, protease, gp120, gp41, virus protein R, viral infectivity factor, virus protein unique, RNA splicing regulator, transactivator protein, negative regulating factor, and virus protein X were −7.02, −6.2, −0.74, −1.22, −8.37, −7.27, −4.97, −8.73, −2.32, −3.24, −5.26, 0.13, −2.75, −1.06, −4.4, and −3.32, respectively.

Ramachandran plots validate the 3D modeled structures of HIV structural and accessory proteins. Ramachandran plot of: (A) capsid (p24), (B) matrix (p17), (C) nucleocapsid (p7), (D) p6, (E) reverse transcriptase (p51/66), (F) invertase (p32), (G) protease (p10), (H) GP120 ()) GP41, (J) virus protein R (p15), (K) viral infectivity factor (p23), (L) virus protein unique (p16), (M) RNA splicing regulator (p19), (N) transactivator protein (p14), (O) negative regulating factor (p27), and (P) virus protein x (p15) proteins. Phi (X-axis) and Psi (Y-axis) represent backbone conformation angles of amino acid residues.
Ramachandran plot statistics of Chi1, Chi2, and BcLPMO proteins.


Local quality estimation graphs comparing the 3D models of expected similarity to the native structures of: (A) capsid (p24), (B) matrix (p17), (C) nucleocapsid (p7), (D) p6, (E) reverse transcriptase (p51/66), (F) invertase (p32), (G) protease (p10), (H) GP120 ()) GP41, (J) virus protein R (p15), (K) viral infectivity factor (p23), (L) virus protein unique (p16), (M) RNA splicing regulator (p19), (N) transactivator protein (p14), (O) negative regulating factor (p27), and (P) virus protein x (p15) proteins of HIV.
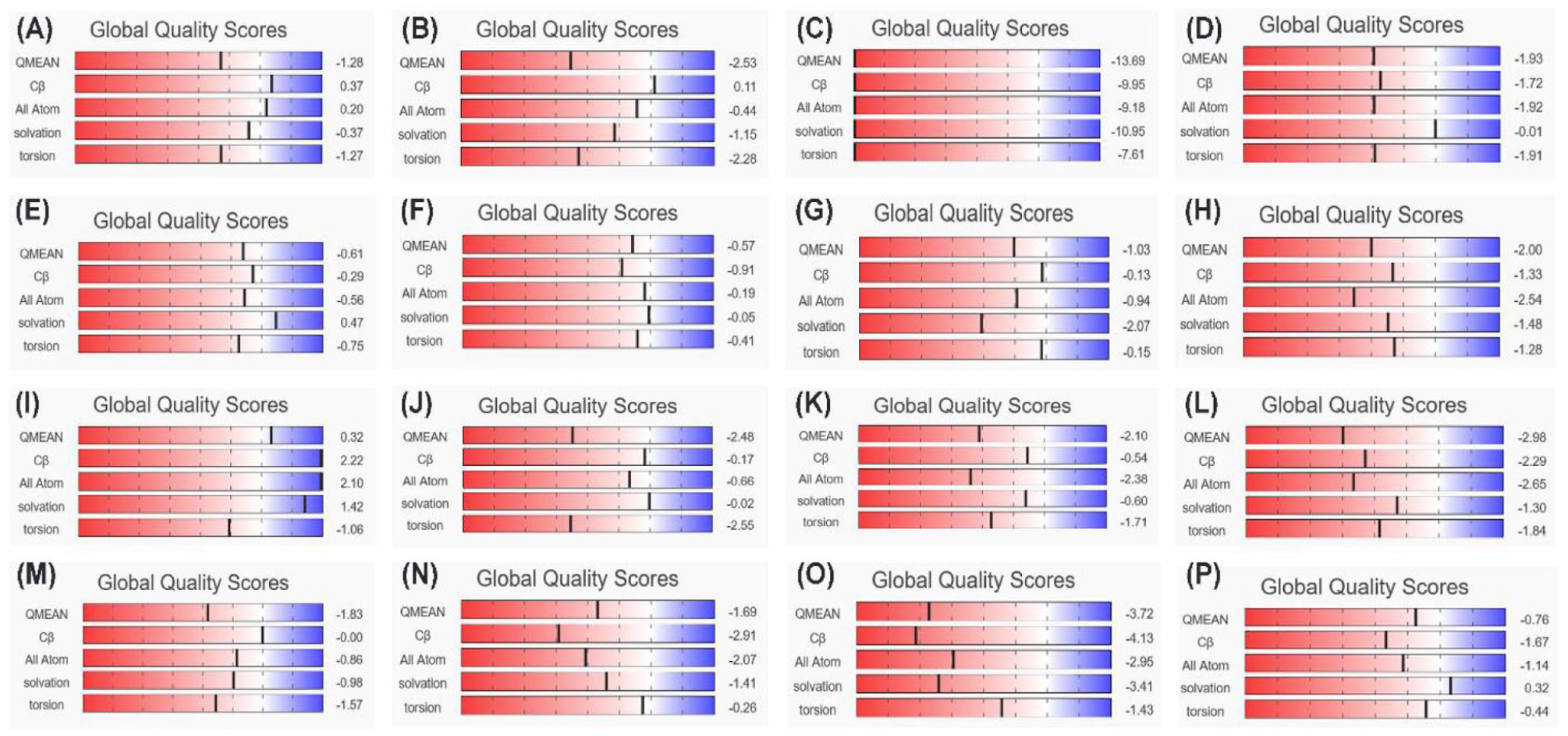
Global quality estimation scores showing the 3D models of entire structures of: (A) capsid (p24), (B) matrix (p17), (C) nucleocapsid (p7), (D) p6, (E) reverse transcriptase (p51/66), (F) invertase (p32), (G) protease (p10), (H) GP120 ()) GP41, (J) virus protein R (p15), (K) viral infectivity factor (p23), (L) virus protein unique (p16), (M) RNA splicing regulator (p19), (N) transactivator protein (p14), (O) negative regulating factor (p27), and (P) virus x (p15) proteins of HIV.

Normalized QMEAN4 score comparing with a non-redundant set of PDB of 3D model structures of: (A) capsid (p24), (B) matrix (p17), (C) nucleocapsid (p7), (D) p6, (E) reverse transcriptase (p51/66), (F) invertase (p32), (G) protease (p10), (H) GP120 ()) GP41, (J) virus protein R (p15), (K) viral infectivity factor (p23), (L) virus protein unique (p16), (M) RNA splicing regulator (p19), (N) transactivator protein (p14), (O) negative regulating factor (p27), and (P) virus protein x (p15) proteins of HIV.

Z-score plot obtained using the ProSA server showing the location of 3D model structures of HIV: (A) capsid (p24), (B) matrix (p17), (C) nucleocapsid (p7), (D) p6, (E) reverse transcriptase (p51/66), (F) invertase (p32), (G) protease (p10), (H) GP120 ()) GP41, (J) virus protein R (p15), (K) viral infectivity factor (p23), (L) virus protein unique (p16), (M) RNA splicing regulator (p19), (N) transactivator protein (p14), (O) negative regulating factor (p27), and (P) virus protein x (p15) proteins in X-ray and NMR structures.
Discussion and Conclusions
Computational protein structure modeling has significantly increased the determination of the 3D structure of proteins as x-ray crystallization and NMR spectroscopy are time-consuming processes and have many difficulties, like purification, crystallization, and low-resolution 3D structures of proteins. 22 Along with structure determination, homology modeling has many other applications, such as structure-based drug designing, mutations analysis, active sites identification, novel ligands designing, substrate-specific modeling, protein-protein molecular docking, molecular simulation, structural refinement at the molecular level, future planning in computational experimentations.39-43 HIV is an infectious disease that attacks the host’s immune cells and weakens the immune system to fight other diseases. HIV encodes 16 distinct proteins, and their 3D structure determination is substantial to understanding their functions in the viral life cycle.44,45 This study identifies the computational 3D modeled of HIV structural and accessory proteins to better understand the structural basis of HIV proteins interacting with host cells and viral replication.
The sequences of HIV capsid (231 bp), matrix (132 bp), nucleocapsid (161 bp), p6 (52 bp), gp120 (455 bp), gp41 (72 bp), reverse transcriptase (259 bp), invertase (288 bp), protease (99 bp), virus protein r (96 bp), viral infectivity factor (192 bp), virus protein unique (81 bp), RNA splicing regulator (116 bp), transactivator protein (86 bp), negative regulating factor (206 bp), and virus protein x (113 bp) proteins were downloaded from UniPort. All the UniPort KB IDs with protein sequences of HIV proteins are mentioned in Table 1. The primary structures were determined by using HIV protein sequences in the ProtParam tool of the Expasy server. In this study, the theoretical pI of gp120 was calculated as 8.53, which was very close to the predicted pI of MN-rgp120 (8.7) and A244-rgp120 (8.4). 46 Similarly, the pI of gp41 was calculated as 4.93, which is acidic, as reported in a previous study. 47 Further, the HIV capsid, nucleocapsid, p6, gp41, virus protein unique, negative regulating factor, and virus protein x showed acidic pI values due to more negative (acidic) amino acid residues (Table 2). While HIV matrix, reverse transcriptase, invertase, protease, gp120, virus protein r, viral infectivity factor, RNA splicing regulator, and transactivator protein had more positive (basic) amino acids, so that showing pI values in a basic range (Table 2).
The HIV proteins, except the transactivator protein, showed 4 secondary structures, that is, alpha helix, extended strand, beta-turn, and random coil, as shown in Table 3. The percentages of secondary structures in HIV proteins vary. Most proteins (nucleocapsid, p6, Reverse transcriptase, invertase, gp120, viral infectivity factor, RNA splicing regulator, transactivator protein, and negative regulating factor) contained more random coils in their secondary structures. While HIV capsid, matrix, gp41, virus protein r, virus protein unique, and virus protein x had more percentage of an alpha helix in their secondary structures. In HIV protease, extended strands were more present than other secondary structures.
For the 3D structure modeling of HIV proteins, templates with maximum sequence identities and similarities were used. Their PDB IDs are mentioned in Table 4. MODELLER software modeled the HIV proteins by using these templates (Figure 1). All the modeled HIV protein structures were validated by Ramachandran plots, local and global quality estimation scores, QMEAN scores, and Z-scores. The Ramachandran plots confirmed the quality of the 3D-modeled structure of the HIV proteins. All the amino acid residues of the capsid, matrix, reverse transcriptase, invertase, protease, gp41, virus protein unique, RNA splicing regulator, transactivator protein, and virus protein x were present in the most favored regions, allowed regions, and generously allowed regions in the Ramachandran plots (Table 5 and Figure 2).
The Ramachandran plot of the 3D modeled structure of HIV-1 capsid protein showed 2 amino acids in the generously allowed region. However, the template (PDB: 5JPA) used to model the HIV-1 capsid protein showed only one amino acid in the generously allowed region of the Ramachandran plot. Similarly, Ramachandran plots of reverse transcriptase, invertase, and protease templates showed 3, 1, and 1 amino acid(s) in the generously allowed region, respectively. While Ramachandran’s plot of modeled predicted structure of reverse transcriptase showed one amino acid in the generously allowed region. Nonetheless, nucleocapsid, p6, gp120, virus protein r, viral infectivity factor, and negative regulating factor had some amino acid residues in the disallowed region of the Ramachandran plots (Table 5 and Figure 2). Additional local quality estimation was used for the estimation of the quality of each residue of the protein. All the 3D modeled protein structures of HIV proteins showed local quality scores above 0.6, except nucleocapsid and p6 proteins (Figure 3). Each residue’s local quality is calculated by comparing its model structure on the x-axis with its native structure on the y-axis. A local quality score below 0.6 is not considered a good 3D-modeled protein structure. 20 A global score determines the overall quality of an entire model. Depending on the alignment and template used to construct the model, the Global Model Quality Estimation can be expressed as a number between 0 and 1. In this number, the denominator is the target sequence coverage. The reliability of the system increases as the number rises.35-38 The global quality score of the 3D modeled HIV proteins showed a QMEAN score above −4.0 except for nucleocapsid protein (QMEAN score −13.69) (Figures 4 and 5). Additionally, the degree of nativeness of 3D modeled structures of HIV proteins with native proteins of similar sizes was calculated by Z-scores. 28 All the 3D modeled structures of HIV proteins showed their native confirmation within the range of the plots, as shown in Figure 6. All the validation results of the HIV 3D modeled proteins confirmed the relatively good quality of predicted proteins which could be helpful in further studies like drug designing.
The present study focused on the HIV structural and accessory proteins 3D structural modeling by using MODELLER. The 3D structures of all the HIV proteins were modeled with maximum sequence similarity and identity protein templates. Further, the predicted structures were validated using a variety of online servers. The obtained 3D modeled structures of HIV proteins might be helpful in further studies to investigate the crystal structures with x-rays crystallography and NMR spectroscopy, proteins motifs and domains, physiochemical properties, new target positions, and drug designing. Moreover, this study predicted the unbounded 3D structures of HIV proteins. However, further studies can predict the bounded structures of HIV proteins, such as HIV RT structure bound with nucleotide RT inhibitors (NRTIs) and nonnucleoside RT inhibitors (NNRTIs). HIV is a dangerous and infectious virus with no cure. Hence, further extensive investigation is still required to find possible treatment in the future.
Footnotes
Declaration of conflicting interests:
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author received no financial support for the research, authorship, and/or publication of this article.