
Abstract
Atypical teratoid rhabdoid tumors (ATRTs) are rare embryonal tumors comprising 1-2% of all pediatric CNS neoplasms. Spinal ATRTs are even more uncommon, accounting for 2% of all reported ATRT cases. Despite their rarity, ATRTs affect young children disproportionately and are characterized by a high malignant potential due to a heterogeneous cellular composition and inactivating mutations in the SMARCB1 (90%) and SMARCA4 (10%) genes. A 15-month-old female presented with a 2-week history of decreased lower extremity movement and new-onset need for assistance with ambulation. MRI lumbar spine revealed a contrast-enhancing intradural mass at the L3-L4 level with iso-intensity on T1 and T2 sequences. The patient subsequently underwent subtotal tumor resection (∼80%) given concerns for maintaining neurological function. Final pathology was consistent with spinal ATRT, and she later underwent adjuvant chemoradiation therapy per ACNS0333 protocol. She has since remained in remission with age-appropriate developmental milestones over the past 2 years. ATRTs should be considered in the differential diagnosis of intradural spinal lesions, especially in the pediatric patient population. Clinical course, presentation, and diagnosis is often delayed due to the rarity of these tumors, but contrasted craniospinal MRI is key for diagnosis and histopathology with IHC staining showing loss of INI is confirmatory. While gross total resection is the goal, maximal safe tumor resection should be prioritized in order to preserve neurological function. Adjuvant chemoradiation following gross total/subtotal resection has been shown to significantly improve overall survival.
Introduction
Atypical teratoid rhabdoid tumors (ATRT) are rare embryonal central nervous system (CNS) tumors that primarily occur in children under 3 years old, and account for 1-2% of all pediatric CNS cancers and 4.4% of CNS tumors in the age group 0-5 years. 1 Prior to 3 years of age, ATRT has a slight male preponderance and recently, it was shown that ATRT is the most common CNS malignancy in children younger than 6 months of age.2,3 Most of these tumors occur intracranially, with 55-75% of these lesions encountered in infratentorial locations and 25-40% located supratentorially, even rarely occurring in the ventricles.1,4 Spinal location for ATRTs is exceedingly rare and has an estimated prevalence of 2% among all CNS ATRTs.1,5
Primary spinal ATRT is a rare subset of ATRT, with very few cases documented in the English-language literature. Often, pediatric patients with spinal ATRT present with rapidly progressive symptoms of pain and myelopathy due to spinal cord compression and early diagnosis and management is key to improve overall outcome.2,6,7 Though the radiological appearance of these rare tumors is non-diagnostic, histological examination of the tissue with the use of immunohistochemical staining is key for making the diagnosis.6,7
ATRTs are characterized by their heterogeneous histology and cellular composition, and consist of varying proportions of rhabdoid, epithelial, mesenchymal, spindled, and primitive neuroectodermal tumor (PNET) cells.8,9 Despite this morphologic heterogeneity, all ATRTs are associated with inactivating bi-allelic mutations in one of two genes, SMARCB1 (90% of reported cases) or SMARCA4 (10% of reported cases), resulting in a loss-of-function mutation in the chromatin-remodeling complex responsible for cell growth inhibition. 10 Together, these mutations and cellular makeup confer a highly malignant potential, with 20-40% of all ATRTs reported to present with leptomeningeal metastasis from their primary source on initial presentation.11,12
Here we report the case of an intradural, extramedullary spinal ATRT found in a 15-month-old girl that responded well to subtotal resection followed by adjuvant chemoradiation therapy with ACNS0333 protocol.
Case presentation
A 15-month. female presented to an outside hospital with a 2-week history of fussiness, decreased lower extremity movement, and new-onset need for assistance with walking. The patient’s prenatal history was unremarkable, with a normal spontaneous vaginal delivery at 37 weeks term without any post-natal complications or history of congenital conditions in the family. Additionally, family history was negative for history of malignancy. On presentation, she was awake and alert, interactive, and demonstrated adequate tone and reflexes. However, she would not stand independently and cried upon lying prone. The patient was diagnosed with herpangina after oral ulcers were noted on physical exam, and work-up for osteomyelitis vs transient synovitis vs septic joint was initiated. Incidentally, an MRI of the pelvis obtained for further workup showed an intradural mass at the L3-L4 level with iso-intensity on T1 and T2 and mild enhancement on post-gadolinium contrast. Urinary retention was also noted on MRI, and a foley catheter was placed for suspected neurogenic bladder before the patient was transferred to the University of Iowa Hospitals and Clinics.
Subsequent total-spine MRI the following day revealed a large, intradural extramedullary mass caudal and ventral to the cauda equina spanning from L1-L4, measuring 13 × 17 × 53 mm3 (Figure 1A-D). The mass showed heterogeneous enhancement and diffusion restriction. On gradient images, the mass exhibited heterogeneous foci of blooming, suggestive of hemorrhagic foci. Given these findings, the initial differential consisted of small round blue cell tumors, such as embryonal tumors or Burkitt Lymphoma. Myxopapillary ependymoma was also considered but thought to be less likely on the differential given the patient’s age. Following preoperative optimization, the patient underwent a posterior approach L1-L4 laminoplasty for intradural mass resection. Following bilateral L1-L4 laminectomy, intraoperative ultrasound was used to ensure that the intradural mass was localized rostrally and caudally within the margins of the bony exposure. The dura was then incised under microscopic visualization and the cauda equina and nerve roots overlying the mass immediately herniated dorsally through the dural and arachnoid defect. These nerve roots were micro-dissected with Penfield 3 and Rhoton 7 micro dissectors to expose a purple, friable mass with a central capsule (Figure 2A-C). Nerve roots were intertwined with the mass both rostrally and caudally. Following central debulking with bipolar cautery and suction, the capsule was mobilized rostrally and separated from the overlying nerve roots. Debulking continued in a caudal to rostral fashion with SSEP and EMG monitoring to avoid conus and nerve root injury. The most rostral aspect of the tumor was noted to be tightly adherent and ventral to the conus with no clear dissection plane identified. At this stage, the frozen specimen sent for pathology resulted and was concerning for a small round blue cell tumor with a differential of sarcoma, lymphoma, or germ cell tumor being considered likely. Due to the possibility of medical management for these differential diagnoses, resection was halted with ∼80% of the tumor resected. Total tumor resection was avoided given concern for neurological injury given the remaining 20% of the tumor was abutting the conus medullaris with no clear dissection plane. Pre-operative MRI of the spine: Sagittal T1 post-gadolinium contrast (A), sagittal T2 (B), axial T1 post-contrast (C), and axial DWI (D) demonstrating an intradural extramedullary mass (orange arrow) caudal and ventral to the conus medullaris (green arrow) at L1-L4 with T1 and T2 Iso-intensity, heterogeneous enhancement, and diffusion restriction (blue arrow). Note the largely distended bladder due to the mass effect and compression on the conus medullaris affecting bladder emptying (blue star). Intraoperative pictures. (A) Dorsal approach posterior L1-4 laminectomy with exposure of underlying dura (caudal is to the right). (B) Following dural opening, Penfield micro-dissectors are used to dissect the cauda equina and nerve roots ventral to the mass exposing a purple, friable mass. (C) Following exposure, the mass was centrally debulked with bipolar cautery and suction. The cauda equina nerve roots can be seen dorsally displaced. (D) After subtotal resection of spinal mass with residual mass ventral to the conus. (E) Expansile duraplasty was performed with Durerepair for dural closure. (F) The L1-L4 spinous processes and laminae were then reattached with KLS martin plates and screws to complete the laminoplasty.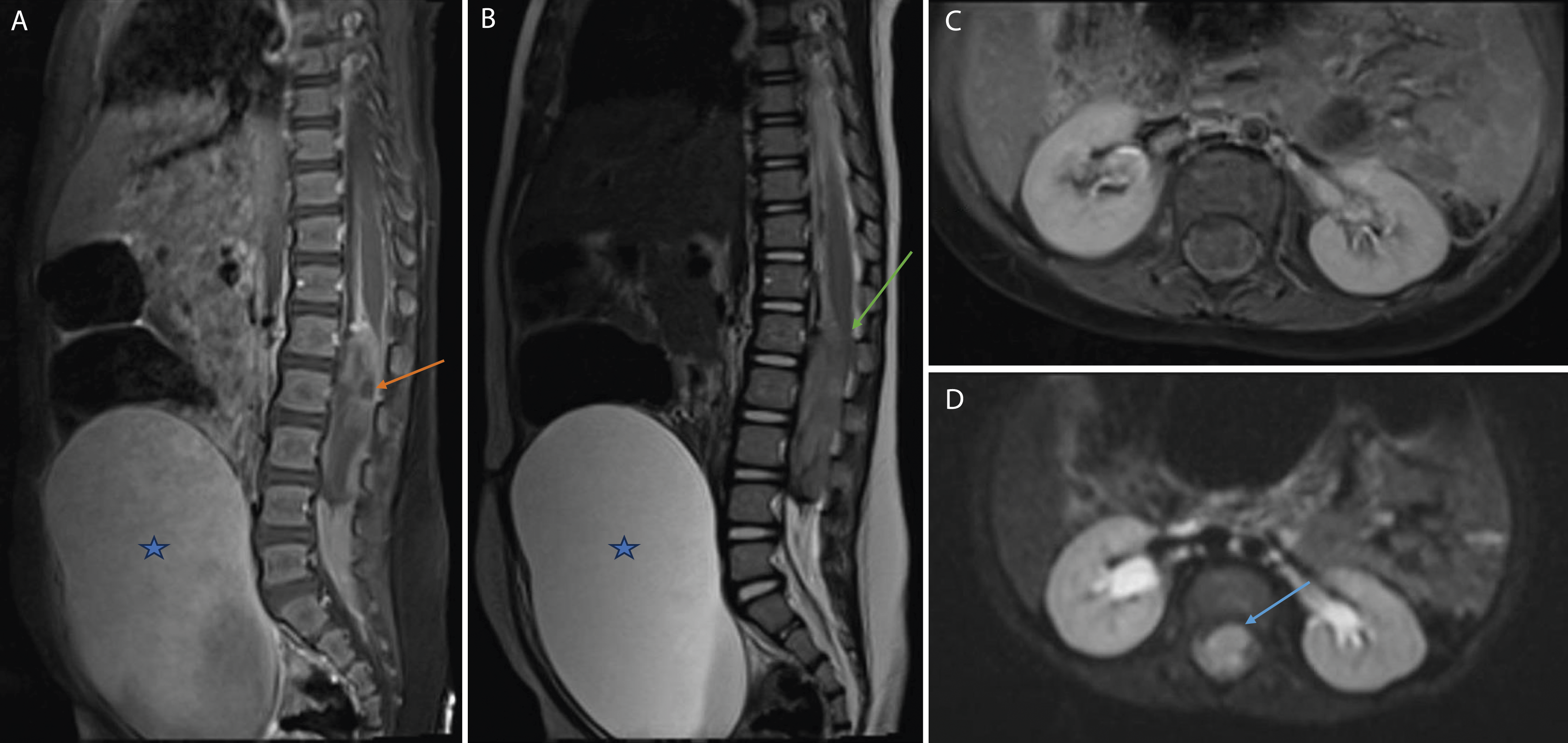

Following satisfactory hemostasis, primary dural closure was attempted but unsuccessful due to continued dorsal herniation of the cauda equina and nerve roots (Figure 2D-F). Expansile duraplasty was then performed with Durepair (TEI Biosciences, Medtronic, Boston, MA) with 4-0 Nurolon sutures and Tisseel (Baxter International Inc., Westlake Village, CA) for dural sealant. Gel foam was placed on the dura, and the L1-L4 spinous processes and laminae were then reattached with KLS martin plates and screws (KLS Martin LLC, Jacksonville, FL). Vancomycin antibiotic powder was applied, and the patient’s fascia and skin were closed using 3-0 nylon in a simple running fashion. The patient tolerated the procedure well with no postoperative complications.
Final histopathology of the tumor demonstrated poorly differentiated and discohesive cells with high nuclear to cytoplasmic ratios, vesicular chromatin, and prominent nucleoli (Figure 3A-D). Immunohistochemistry revealed immunoreactivity for epithelial membrane antigen (EMA), pankeratin, neurofilament, and smooth muscle-specific actin (SMSA) with loss of INI-1/SMARCB1, confirming the diagnosis of ATRT. The patient was initiated on 2 cycles of multiagent chemotherapy using cisplatin, cyclophosphamide, vincristine, etoposide, and methotrexate, followed by 3-course of high-dose chemotherapy with proton-based radiation treatment and autologous peripheral blood stem cell transplant per ACNS0333 guideline. Post-operatively, the patient remained neurologically stable with continued improvement of her lower extremity weakness throughout her hospital stay. The patient was discharged on post-operative day 27 following completion of her first chemotherapy regimen. She was noted to be ambulatory with moderate assistance. 6 weeks after surgery, post-operative MRI spine confirmed residual tumor along the conus with no new enhancing spinal lesions (Figure 4A-D). The patient continued with 3 cycles of chemotherapeutic regimen per ACNS0333 guidelines over a 6-month period. At her most recent follow-up over 2 years after surgery, the patient’s cancer remains in remission with age-appropriate developmental milestones (Figure 5). (A) H&E, 400× magnification: Intraoperative touch prep demonstrating dis-cohesive tumor cells with irregular nuclear contours, vesicular chromatin, and scant cytoplasm. (B) H&E, 400× magnification: Sheets of tumor cells with pleomorphic nuclei, prominent nucleoli, and scant cytoplasm. (C) H&E, 200× magnification: Small-round blue cell neoplasm with broad regions of necrosis. (D) INI1 immunohistochemical stain, 200x: Loss of nuclear expression of INI1 within tumor cells with preservation of expression within internal control (endothelial cells). Post-operative MRI of the spine: Sagittal T1 post-gadolinium contrast (A), sagittal T2 (B), axial T1 post-contrast (C), and axial T2 (D) demonstrating persistent intradural extramedullary mass postoperatively (orange arrow) after incomplete resection with expected internal hemorrhagic changes and decreased mass effect on the conus medullaris (green arrow). Repeat MRI spine 5 months post-adjuvant chemoradiation: Sagittal T1 post-gadolinium contrast (A), sagittal T2 (B), axial DWI (C), and axial T2 (D) demonstrating reduced tumor size (orange arrow) without new enhancing lesions and decompressed conus medullaris (green arrow) and cauda equina (blue arrow).


Discussion
Atypical teratoid rhabdoid tumors (ATRTs) are rare embryonal tumors, representing only 1-2% of all pediatric CNS tumors.13,14 ATRTs originating primarily within the spine are estimated to account for less than 2% of all CNS ATRTs. 15 Despite their rarity, ATRTs disproportionately affect younger children; 40-50% percent of CNS malignancies in children aged less than 1 are ATRTs, and it is imperative to consider ATRTs in the differential diagnosis of pediatric craniospinal neoplasms. 14 Our patient initially presented to an outside hospital with lower extremity weakness and was referred to the University of Iowa Hospitals and Clinics for further evaluation after a lumbar lesion was discovered incidentally on MRI. Subsequent spinal imaging prompted tumor workup and evaluation, culminating in intradural mass resection in conjunction with ACNS0333 chemotherapy and adjuvant proton-beam radiation treatment.
Histopathology/molecular features
Rhabdoid cell tumors were originally described in the context of renal tumors; however, in 1996 a seminal study established the presence of rhabdoid cell tumors within the central nervous system as being distinct from other lesions, such as medulloblastomas or primitive neuroectodermal tumors.16,17 In addition to rhabdoid cells, the researchers identified epithelial, mesenchymal, and large and small cells consistent with a teratoid morphology, from which the tumor derives its name. 8 Since then, spindle cells and sickle-shaped cells have also been identified on histologic sectioning. 9
Despite this cellular diversity, all ATRTs are associated with mutations in the SMARCB1 or SMARCA4 genes, causing a loss-of-function protein deactivation. 10 These genes encode proteins involved in the SWI/SNF chromatin remodeling complex, named SMARCB1 and SMARCA4. 10 The SWI/SNF complex is responsible for several cellular processes including DNA mismatch repair and chromatin structure regulation. 18 Mutations in these key proteins result in decreased strand fidelity due to faulty mismatch repair, as well as uncontrolled cell growth resulting from dysregulated chromatin remodeling and increased DNA replication with looser chromatin structure. 19 Finally, mutations in the SMARCB1 and SMARCA4 are associated with variable levels of DNA hypomethylation, suggesting epigenetic alterations of cell growth regulation. 10
Although bi-allelic SMARCB1 loss is the critical genetic event in the vast majority of ATRTs, despite this genetic homogeneity, ATRTs are an epigenetically diverse group of tumors with distinct enhancer landscapes.20-22 At least three molecular subtypes of ATRTs exist, each with unique targetable pathways and potential therapeutic vulnerabilities: ATRT‐TYR, ATRT‐SHH, and ATRT‐MYC.2-22 ATRT-TYR and ATRT-MYC subtypes display a critical dependence on receptor tyrosine kinase pathways. The tyrosine kinase inhibitors (TKIs) dasatinib and nilotinib display selective toxicity to ATRT-MYC and TYR cell lines, and dasatinib significantly improved survival in an intracranial orthotopic xenograft model. 23 ATRT-SHH tumors appear to be more critically dependent on multiple targetable epigenetic regulators for survival like EZH2 and inhibition with an EZH2 inhibitor (UNC1999) has been shown to be selectively toxic to cell lines from this subgroup. 23 A phase-I trial using the EZH2 inhibitor (tazemetostast) for relapsed or refractory SMARCB1-deficient tumors is currently ongoing (NCT02601937). All these ongoing clinical trials and laboratory research focused on translating these exciting new insights into efficacious therapeutics, will hopefully improve the outcome of this devastating disease.In terms of histopathology, intraoperative frozen pathology obtained from our patient was notable for a small round blue cell tumor with differentials of sarcoma, lymphoma and germ cell tumor considered more likely given the patient’s age and tumor location. However, due to the histological similarity between other embryonal tumors and ATRTs, further molecular and histological work-up was pursued. Our patient’s tumor molecular profile disclosed a SMARCB1 pathogenic variant at a 50% allelic fraction and loss of INI-1 expression, consistent with one of the two ATRT mutations. Immunohistochemistry revealed EMA-1, pankeratin, neurofilament, and SMSA positive staining, further supporting the diagnosis of spinal ATRT by identifying rhabdoid, epithelial, and PNET cells.24,25 Most importantly, IHC was absent for SMARCB1 protein, allowing a definite diagnosis of ATRT to be made. 26
Clinical and radiological features
Summary of published cases of pediatric spinal ATRT in the English literature.

Legend: — = not reported; m = months, yr = years; ED = extradural, ID = intradural, EM = extramedullary, IM = intramedullary, IT = intracranial; Y = yes, N = no; ASCR = autologous stem cell rescue, SRS = stereotactic radiosurgery, DOD = died of disease, NED = no evidence of recurrence.
Treatment and long-term outcomes
The prognosis of ATRTs is highly variable depending on tumor location, clinical presentation, and presence of metastases. A 2022 systematic review by Egiz et al of 39 retrospective studies found the median overall survival rate of pediatric ATRT diagnoses to be 29 months. 72 ATRTs of spinal origin, as was found in our patient, demonstrate a significantly lower median survival rate compared to those that are cranial in origin. For primary tumors, surgical resection is a mainstay of treatment and prognosis may correlate with the extent of the tumor that can be safely resected. 72 Egiz et al found that complete tumor resection increased the median survival rate to 4.2 years as compared to .5-.9 years in patients with partial or subtotal resection. Adjuvant therapy is also recommended, with chemotherapy, combination chemotherapy and photon or proton-based radiation treatments, all having been utilized for multimodal management. 73 A combination of chemotherapy and radiation is most common with a median survival rate of 8.842 years. 72 Prognosis worsened to a median survival rate of 4.942 years in patients who receive proton-based radiation therapy, and only .833 years in patients who receive chemotherapy as a lone adjuvant to surgical resection. 72 This data suggests that maximal tumor resection alongside chemotherapy and radiation may provide optimal patient outcomes. However, due to the lack of a standard treatment protocol, various multimodal regimens are still being explored through clinical trials as seen in our patient’s case.
Our patient underwent surgical resection followed by high-dose chemotherapy with peripheral blood stem cell rescue as outlined by the ACNS0333 clinical trial protocol, followed by 6 weeks of proton-based radiation therapy. 74 Given her young age, proton-based radiation treatment was chosen to reduce the risk of late radiation treatment related effects. Proton-based radiation therapy has gained wide acceptance in the pediatric oncology community as a modality to minimize the toxicities associated with photon-based radiation treatments.75,76 Given the physical properties of the proton beam, it allows for conformation of the radiation dose more strictly to the volume of interest, avoiding critical organs with greater normal tissue sparing than occurs with photons and is a theoretical advantage because of the reduction of integral dose to normal tissues (ex. conus medullaris in our patient).75,76 This ability of protons to spare normal tissues beyond a specified depth results in lower or no dose to surrounding organs with subsequent reduced toxicities.75,76
Currently, our patient remains in remission 27 months from her initial diagnosis. We demonstrate that sub-total surgical resection followed by aggressive adjuvant therapy can allow patients to achieve remission and continue development appropriately, with minimal neurologic impairment.
Supplemental Material
Supplemental Material - Clinical diagnostic and radiographic features of primary spinal atypical teratoid rhabdoid tumor in a pediatric patient: A case report and review of the literature
Supplemental Material for Clinical diagnostic and radiographic features of primary spinal atypical teratoid rhabdoid tumor in a pediatric patient: A case report and review of the literature by Hashim Syed, Nahom Teferi, Alec Hanson, Meron Challa, Kathryn Eschbacher and Patrick Hitchon in Journal of Central Nervous System Disease
Ethical statement
Ethics approval and consent to participate
The authors of this study confirm that written informed consent was obtained from the patient’s parental guardians for participation in this study. The University of Iowa Hospital institutional review board approved all aspects of this study under IRB # 201902751.
Consent for publication
The authors of this study confirm that a written informed consent for publication of the patient's medical images and data was obtained from the patient’s parents.
Footnotes
Author contributions
Conception and design: Nahom Teferi. Acquisition of data: Hashim Syed, Alec Hanson. Analysis and interpretation of data: Hashim Syed, Alec Hanson Drafting of the article: Hashim Syed, Alec Hanson. Critically revising the article: Nahom Teferi, Meron Challa, Kathryn Eschbacher, Patrick Hitchon. Approved the final version of the manuscript on behalf of all authors: Nahom Teferi.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material:
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.