
Abstract
Huntington’s disease (HD) is an autosomal neurodegenerative disease that is characterized by an excessive number of CAG trinucleotide repeats within the huntingtin gene (HTT). HD patients can present with a variety of symptoms including chorea, behavioural and psychiatric abnormalities and cognitive decline. Each patient has a unique combination of symptoms, and although these can be managed using a range of medications and non-drug treatments there is currently no cure for the disease. Current therapies prescribed for HD can be categorized by the symptom they treat. These categories include chorea medication, antipsychotic medication, antidepressants, mood stabilizing medication as well as non-drug therapies. Fortunately, there are also many new HD therapeutics currently undergoing clinical trials that target the disease at its origin; lowering the levels of mutant huntingtin protein (mHTT). Currently, much attention is being directed to antisense oligonucleotide (ASO) therapies, which bind to pre-RNA or mRNA and can alter protein expression via RNA degradation, blocking translation or splice modulation. Other potential therapies in clinical development include RNA interference (RNAi) therapies, RNA targeting small molecule therapies, stem cell therapies, antibody therapies, non-RNA targeting small molecule therapies and neuroinflammation targeted therapies. Potential therapies in pre-clinical development include Zinc-Finger Protein (ZFP) therapies, transcription activator-like effector nuclease (TALEN) therapies and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated system (Cas) therapies. This comprehensive review aims to discuss the efficacy of current HD treatments and explore the clinical trial progress of emerging potential HD therapeutics.
Keywords
Introduction
HD is an autosomal dominant neurodegenerative disease that currently has no approved cure. HD is characterized by an increased number of CAG trinucleotide repeats in the huntingtin gene (HTT) on the short arm of chromosome 4. A higher number of CAG repeats in the huntingtin gene is directly associated with a greater probability of HD onset. Individuals with greater than 36 CAG repeats have an increased risk of HD, while patients with over 40 repeats are predestined to experience disease onset within their life span.1-5 The HTT gene which possesses the CAG repeats is transcribed into the huntingtin protein (HTT).
6
A high number of CAG repeats results in a dysfunctional mHTT protein, forming protein aggregates within the neurons and other cell types.
7
These aggregates, among other mechanisms, are hypothesized to cause mitochondrial dysfunction, neuronal stress, excitotoxicity and neuroinflammation (Figure 1).8-12 The onset of HD typically occurs at middle age, however, HD symptoms can develop at any time during the lifespan.13,14 Worldwide, the prevalence of HD is 2.71 per 100,000, however, this number increases to 5.70 per 100,000 in westernised countries such as Europe, the United States and Australia.
15
This has been postulated to be the result of a higher frequency of the mutant HD gene in western haplotypes.
16
Mechanisms of Huntington’s disease pathogenesis.
A variety of symptoms have been recorded with the majority resulting in the deterioration of cognitive ability, motor function, a change in behaviour or a combination of these symptoms.17,18 At present, there are no approved treatments that directly target the mutated HTT gene or corresponding mHTT aggregates. Current approved HD drugs address the range of symptoms that arise as a consequence of the disease, but they do not address the disease at its origin. 19 Each patient’s symptoms can be unique, hence there is no standardized treatment with medication prescribed on a case-by-case basis and an extensive range of medications used to treat each of the unique symptoms.13,20 Side effects from medications used to treat HD can also affect each patient differently. 18
Most HD symptoms can be classified under motor, cognitive or behavioural categories.14,18 Motor symptoms include chorea during the early and mid-stages of disease onset, however, in later stages of disease progression movements become slower (bradykinetic) and less frequent (hypokinetic). 21 Alternatively, behavioural symptoms include depression, apathy and anxiety.22,23 In 1988, a survey of 2835 HD patients discovered that 40% of patients had depression at the time of the study, while 50% had sought clinical help for depression in the past. 24 Other less common psychiatric symptoms include obsessive-compulsive behaviour, psychosis and aggression. Memory deficits and retrogression of thought processing and organization can arise as cognitive symptoms.21,24 Other symptoms can also present, such as unintentional weight loss and sleep difficulties. 14
The field of potential future HD therapies is advancing rapidly and holds an exciting future. Currently, attention is particularly focussed on lowering mHTT through RNA and DNA targeting therapies. 25 Of these, antisense oligonucleotides are the most rapidly advancing potential therapies. 26 Currently, Tominersen has one of the largest phase 3 clinical trials, and results from this trial were expected in 2022.27,28 However, the dosing in this trial has been prematurely discontinued and the results so far are being assessed. 29 Most RNA and DNA targeting therapies are delivered invasively via intrathecal or intracranial injection. 30 Furthermore, DNA based therapies are all in pre-clinical stages and have added ethical complications due to their potential to edit germline DNA. 31 Other less invasive therapies are therefore being investigated, including stem cell therapies, antibody therapies (both mostly delivered intravenously) and small molecule therapies (delivered orally).25,32,33 All of these concepts hold promise and have their own strengths and weaknesses. Many are in clinical trials with manifest HD participants, 27 however, there is still significant development of these drugs needed until new therapies are approved and available to HD patients. Accessible new therapies will likely take many years to eventuate. 34 This comprehensive review will aim to compare the relative effectiveness of currently approved medications for HD and explore new treatment possibilities, with a special focus on those undergoing clinical trials. We intend to provide a broad overview of the field of current and potential future HD therapies as a forefront resource, rather than focussing on specific therapy types as many previous reviews have done. 35 A particular emphasis is placed on currently researched therapies, but therapies investigated in the past 5 to 10 years were included if they had been comprehensively studied or discussed in their subfield.
Current Therapeutic Options for Huntington’s Disease
Chorea Medications
Currently used medications for HD treatment.

Tetrabenazine
Tetrabenazine (TBZ) was the first drug approved to treat Huntington’s disease-associated chorea. 41 Although the direct link between its mechanism of action and overall pharmacodynamic effect of reducing chorea is not fully understood, its pharmacological action is thought to selectively deplete central monoamines from the nerve terminal by reversibly inhibiting the human vesicular monoamine transporter 2 (VMAT2). 42 To understand the effectiveness of the drug, a randomized control trial was completed by the Huntington’s Study Group where 84 patients received TBZ (n = 54) or placebo (n = 30) over 12 weeks. 43 The dose was gradually increased over the first 7 weeks to either a maximum of 100 mg/day, until desired anti-chorea effects were achieved, or until side effects prevented continued participation of the patient. The outcome was measured via a change in chorea score of the Unified Huntington’s Disease Rating Scale (UHDRS). 44 Following the 12 weeks, it was concluded that TBZ significantly reduced chorea severity by 5.0 UHDRS units relative to only 1.5 UHDRS units in the placebo treatment. Four subjects did not complete the treatment due to adverse side effects. Following on from this, another 15-year longitudinal study examined patients taking TBZ and found that 17.2% of HD patients experienced adverse side effects including drowsiness, parkinsonism, depression, insomnia, anxiety and akathisia. However, the majority of these side effects were eliminated with a reduction in dose. 45
Deutetrabenazine
A similar medication to TBZ used to treat HD-associated chorea is deutetrabenazine (DBZ). DBZ is an isotopic isomer of TBZ, where 6 hydrogen atoms have been replaced by deuterium atoms. Therefore, DBZ has the same chemical function as TBZ, however, the deuterium atoms extend the half-life of the compound, in turn allowing for less frequent administration.20,46 A clinical trial, completed by Frank et al. (2016) randomized 90 HD patients between being administered DBZ (n = 45) or a placebo (n = 45) over a 12- week period (NCT01795859). Patient chorea scores were both measured at baseline and following the treatment intervention. HD patients receiving DBZ all experienced significant improvements in their chorea scores at the end of the 12 weeks relative to the control patients. 47
Both TBZ and DBZ prescriptions are labelled with warnings about the side effects of depression and suicidality. 48 However, individuals with HD taking DBZ had a significantly lower risk of developing neuropsychiatric adverse outcomes (agitation, depression, drowsiness, insomnia and parkinsonism) relative to individuals with HD taking tetrabenazine. 49 Since the induction of the drug in a clinical setting various studies have been completed analysing the link between suicidality and TBZ/DBZ. Experiments from both Dorsney et al. (2013), and Shultz et al. (2018), both found that TBZ use was not associated with an increased incidence of depression or suicidality.50,51 The contrast in findings could be due to the unaccounted increase in depression and suicidality from the HD disease alone. 24 It is known that depression significantly increases in HD patients with 10% to 25% attempting suicide at least once.24,52 These studies highlight the need for further research to be conducted to understand whether the increase in depression and suicidality is a result of TBZ/DBZ or if HD patients already have an underlying susceptibility to these symptoms.
Dopamine antagonists (Antipsychotics or neuroleptics)
Dopamine antagonists are used as second-line agents for chorea relief. These medications are primarily used for the treatment of psychosis, however, they also help to relieve chorea symptoms due to their interaction with the D2 receptor. Specific medications that antagonise dopamine are discussed in further detail below under antipsychotic medication53-55.
Anti-glutamatergics
There has also been some research into the role that glutamate excitotoxicity plays in HD-associated chorea. Hence, medications such as amantadine and riluzole have been trialed as anti-glutamatergics to reduce chorea symptoms. 56
Amantadine
Amantadine is a non-competitive N-methyl-D-aspartate (NMDA) receptor antagonist. However, it is hypothesized to affect the dopaminergic system and is considered the most effective agent to treat levodopa-induced dyskinesia in patients with Parkinson’s disease. 57 Verhagen Metman et al. trialed Amantadine to treat HD-associated chorea. To evaluate Amantidine’s effects, 24 HD patients entered a double blinded study where the treatment group received 400 mg/day of Amantadine. Over a 2-week treatment period, chorea scores were lower in the treatment group, with the median decrease in extremity chorea at rest being 36% (P = .04). 58
Riluzole
Riluzole is another anti-glutamatergic that has also been trialed to treat Huntington’s associated chorea. One study by Rosas et al. treated 8 HD patients with 50 mg of riluzole twice a day over 6 weeks. They found that the patients’ chorea rating score improved by 35% (P = .013) and regressed back to their original scores once discontinued (P = .026). 59 Alternatively, a similar study treated 9 HD patients with an identical dosing regime, but over 12 months. They discovered that patients had improved chorea scores at 3 months, however, this was sustained at 12 months. 60 Therefore, the long-term efficacy of riluzole to treat HD-specific chorea is still in question.
Antipsychotic Medication
Olanzapine
Olanzapine is the second most commonly prescribed drug for HD 20 (Table 1). Olanzapine is traditionally used as an antipsychotic drug that is primarily used to treat symptoms associated with schizophrenia. 61 Olanzapine is now prescribed to treat many HD patients expressing behavioural symptomology. A study on the short-term effects of olanzapine with HD patients (n = 11) found that 6 months following administration their behavioural assessment scores within the UHDRS significantly improved. 62 Chorea scores also improved in these patients, however, were not significant. This suggests that olanzapine may be a more suitable medication for HD patients with behavioural symptoms, while TBZ/DBZ would better address HD-associated chorea. Patients who present with multiple symptom groups are often prescribed multiple medications, taking into account possible negative drug-drug interactions. 21 This example again illustrates that there is no universal approach to treating HD and highlights the need for each patient to be assessed individually.
Olanzapine inhibits dopamine receptors (D1, D2 and D4), serotonin receptors (5-HT2A, 5-HT2C), histamine receptors (H1) as well as a1-adrenergic and muscarinic receptors. 54 Unlike the majority of other antipsychotic medications, olanzapine has a higher affinity for 5-HT receptors compared to D2 receptors, therefore, being classed as an atypical antipsychotic. 54
Side effects of this medication include excess weight gain and dyslipidemia. This has been observed in rat studies as well as clinical trials.63,64 To overcome this, it is important to take into account the patient’s current metabolic health before prescribing the medication. A study by Ball et al., (2001) found that by placing olanzapine patients on a weight loss program they could mitigate some of the weight gain effects. However, sustained weight management was only observed in males. 65
Risperidone
Another traditionally used antipsychotic medication that is prescribed to HD patients is risperidone. 20 Similar to olanzapine, risperidone has antagonistic properties, selectively inhibiting serotonin-S2 (5-HT) and dopamine-D2 receptors 55 and is commonly used to treat schizophrenia. 66 Risperidone is an atypical antipsychotic drug. An atypical antipsychotic is a drug that has a higher binding affinity to 5-HT receptors (e.g. olanzapine), while alternatively, a typical antipsychotic has a higher binding affinity to D2 receptors. 67 However, it is important to note that while risperidone is classified as an atypical antipsychotic drug it binds to both 5-HT2 and D2 receptors. 68
Research completed by Duff et al. (2008) administered risperidone to 17 HD patients over 14 months. They found that patients taking risperidone medication had significantly improved behavioural and psychiatric assessment scores compared to controls (n = 12). 69 Additionally, motor symptoms of the treatment group stabilized while the control group’s motor symptoms deteriorated 69 (NCT04201834). This suggests that risperidone may have dual benefits for both the treatment of motor and behavioural symptoms associated with HD. However, further evidence supporting this dual benefit of risperidone is minimal, therefore, larger randomized control trials must to be completed in the future to understand the drug’s full efficacy.
A randomized control trial was completed by Conley & Mahmoud, comparing the relative efficacy of risperidone and olanzapine at reducing adverse cognitive and mood behaviours in Schizophrenic patients (n = 377) over 8 weeks. 70 Both treatments were efficacious and well tolerated by patients. The only difference was that a greater proportion of patients taking olanzapine (27%) experienced a significant increase in body weight percentage relative to those taking risperidone (12%). 70 This suggests that weight gain is an area of concern when prescribing antipsychotic drugs to HD patients, and highlights risperidone as a potentially more suitable option.
Antidepressants
Approximately 40% of HD patients experience partial or ongoing HD-associated depression and 1 in 10 attempt suicide post-disease diagnosis.24,52,71 Antidepressant drugs are often prescribed to HD patients suffering from depression, however, these do not address other psychotic symptoms nor the Huntington’s associated chorea. The majority of antidepressants used to treat HD-associated depression are selective serotonin reuptake inhibitors (SSRIs) (Table 1). SSRIs preferentially inhibit the reuptake of 5-HT into the presynaptic nerve terminal. This increases the concentration of 5-HT in the extracellular environment, increasing the amount of 5-HT available to bind to receptors on the post-synaptic cleft,72-74 which leads to desensitization of the receptors. Desensitization of serotonin receptors may contribute to the therapeutic actions of SSRIs and also account for the development of tolerance to acute side effects of these drugs. SSRIs might result in strong but slow disinhibition of 5-HT neurotransmission. Treatment with antidepressants also causes a downregulation of 5-HT2 receptors.75-77
SSRIs also have several undesirable side effects such as sexual dysfunction and weight gain. 78 These symptoms typically arise following a month of treatment and can be prevented if diagnosed early. 78 Some studies have also found an increased risk of suicide in adolescents taking SSRIs, however, this may be less of an issue in HD patients as HD onset does not usually occur until middle age. This same risk factor has not been observed in adults using SSRI medication.79,80 Overall, the literature suggests an inadequate knowledge to properly manage how antidepressants are prescribed to HD patients. 81 Further research is required to understand which SRRI’s are most suitable for HD patients and whether these are the best medication to address the high rates of suicidality.
Citalopram
A common SSRI that is prescribed to HD patients is citalopram. A randomized control trial by Beglinger et al., (2014) administered HD patients with citalopram and measured any relative changes in executive function. Thirty-three adult HD patients were either administered with 20 mg of citalopram or a placebo pill daily over 20 weeks. Although there were no significant changes in executive function, patients showed a significant improvement in depression symptoms on the Hamilton Depression Rating Scale (HAM-D). 82
Fluoxetine
A second SSRI that is prescribed to HD patients is fluoxetine. This drug is used primarily to treat HD-associated unipolar depression. 21 One case study found that 2 non-depressed HD patients who took fluoxetine experienced reduced obsessive compulsive disorder (OCD) symptoms and mood improvements. 83 Fluoxetine acts to reduce depression in the short-term, however, there are limited clinical trials examining its effectiveness in HD patients with a need for further studies becoming evident. 84
Sertraline
Another SSRI that is often prescribed to HD patients is sertraline. This medication not only reduces depression but has also been recorded to cause a complete cessation of aggression and OCD symptoms associated with HD.85,86
Mood Stabilizing Medications
HD patients with behavioural symptoms can also present with symptoms such as OCD, aggression and bipolar disorder. To stabilize these mood disorders, patients can be prescribed a range of anticonvulsants such as sodium valproate, lamtringine and carbamazepine. 21 Anticonvulsants have been reported to be effective at minimizing the severity of these symptoms in numerous studies.87-92
Traditionally, anticonvulsants were commonly used for treating seizures. 93 Seizures are caused by synchronized neuron populations firing abnormally and excessively.94,95 Anticonvulsants reduce this synchrony via 3 main mechanisms: (1) modulation of voltage-gated ion channels; (2) upregulation of inhibitory neurotransmitters; (3) downregulation of excitatory neurotransmitters. 96 However, anticonvulsants like sodium valproate and carbamazepine are also involved in longer term mechanisms such as modulating cellular plasticity and resilience. This is achieved through regulation of inositol biosynthesis (MIP synthase), cyclic adenosine monophosphate (c-AMP) response element binding protein, brain-derived neurotrophic factor (BDNF), mitogen-activated protein kinases and arachidonic acid pathway regulation, among others. 97 Furthermore, sodium valproate inhibits histone deacetylase, creating long-term gene expression changes thought to produce long-term mood stabilizing effects. 98
The exact pharmacodynamics of anticonvulsants is specific to each drug and can include several of the aforementioned mechanisms. 96 Nonetheless, all drugs have similar effects on stabilizing mood in HD patients.21,99-101
Lamotrigine
Lamotrigine also blocks voltage-gated sodium ion channels, however, it also suppresses excitatory neurotransmitters glutamate and aspartate. 102 Although lamotrigine is used as a mood stabilizer in HD, some studies suggest its suppression of excitatory neurotransmitters may reduce neuron excitotoxicity, thus, mitigating one of the pathological symptoms in HD.21,103 A double blind, placebo-controlled study was completed on 64 early diagnosed HD patients (<5 years) over 30 months. The effectiveness of treatment was analysed using a quantified neurological examination and a set of cognitive and motor tests. Lamotrigine did not significantly slow the progression of HD over the 30 months. Therefore, although beneficial against 1 symptom, lamotrigine does not possess neuroprotective attributes.103,104
Carbamazepine
Carbamazepine’s mechanism of action is solely through the blockade of voltage-gated sodium ion channels. However, the exact pharmacodynamics of the drug has not been fully elucidated and there is still some debate within the literature around this.105,106 Although, carbamazepine is prescribed as a mood stabilizing drug for HD, no clinical research was found on its specific benefits to HD patients. 21
Anticonvulsant drugs exhibit a number of side effects. General side effects can include hypersensitivity reactions, blood dyscrasias, dizziness, gastrointestinal disturbances, depression and hyponatremia. 21 Aside from these side effects, carbamazepine also has the potential to cause serious skin conditions such as Stevens-Johnson syndrome or toxic epidermal necrolysis. 107 This could suggest why carbamazepine is prescribed less often than other anticonvulsants. 21
Drug treatment can be beneficial to HD patients, although prescribed drugs should be based on concurrent symptoms as each case is unique. Patients are required to be closely monitored when taking medications and management should be patient specific. Additionally, the side effects of each drug mentioned also need to be carefully recorded and ensured they are balanced with their benefits.
Non-Invasive Strategies and Life-Style Adaptations
The overall treatment of HD requires a holistic approach to health, involving a range of health professionals. 108 Non-invasive strategies and life-style adaptations can also play a vital role in the patient’s management of HD. Chorea is the primary symptom that can severely impair balance and gait. Physiotherapists can support patients by extending the proper function of these actions as well as assessing when walking aids or wheelchairs are required.109,110 Similarly, occupational therapists can assist HD patients through completing home assessments and installing bathroom adaptions or handrails so patients can prolong living in the comfort of their own home. 111
Patients who exhibit motor symptoms can also develop speech impairments such as hyperkinetic dysarthria. Hyperkinetic dysarthria involves involuntary muscle movements in the mouth, throat and respiratory system, resulting in voice hoarseness and abnormal prosody. 112 For these cases, patients can receive speech and language therapy to optimize their verbal communication for as long as possible. If a patient presents with an inability to speak, speech and language therapists can provide communication charts or electronic communicating devices, enabling patients to continue conversing with friends and family.113,114
Malnutrition is another major issue for HD patients, with the majority having a lower than average body weight. 115 Dieticians can help patients in these circumstances, providing them with diet plans and methods to overcome the chorea-induced eating deficits such as blending foods or providing liquid food substitutes. 115 A Percutaneous Endoscopic Gastronomy (PEG) may be performed for some patients in which swallowing is severely affected 116
To alleviate the psychological and cognitive symptoms associated with HD, sessions with a psychologist can be used alongside the medications provided. This may not only improve the patient’s mental health, but also can be used to monitor how the patient is responding to each medication they are receiving. 21
Surgical Treatments
An alternative surgical treatment is deep brain stimulation.117,118 This intervention is primarily used to improve chorea symptoms.119,120 A study from Gonzalez et al. (2014), found deep brain stimulation to be effective at reducing chorea in pharmacologically resistant chorea HD patients. It did not, however, reduce dystonia or bradykinesia. 121 As pharmacological resistant HD is rare, the use of this intervention is rare. The infrequent use of this treatment can also be attributed to the additional risks associated with the invasive procedure. 122
Overall, treatment of HD should not solely be the clinician’s responsibility and the medications they prescribe. For optimal treatment, a range of health care professionals should be involved with an extensive network of communication between them. 123
Potential Future Therapeutic Options for Huntington’s Disease
Antisense Oligonucleotide (ASO) Therapies
Overview of potential future HD treatments.

aAAV.shHD2.1 is also known as AAV.shHD2.

Tominersen and WVE-120101/WVE-120102 therapy development timeline.
Tominersen, WVE-120101 and WVE-120102 function by complementary base pairing to target pre-mRNA, inducing RNase H1-mediated degradation. They reduce mHTT mRNA levels, therefore, lowering mHTT protein synthesis. Administration occurs via intrathecal injection into the cerebrospinal fluid (CSF) via lumbar puncture, allowing distribution to the central nervous system.125,126
Tominersen
Hoffman-La Roche’s RO7234292 or Tominersen (previously called IONIS-HTTRx/RG6042; IONIS Pharmaceuticals) is an ASO that binds to wild type HTT (wtHTT) and mHTT mRNA, inducing degradation. 126 It is currently the most developed HD ASO therapeutic. 27
Tominersen distributed well in mice and non-human primate brains during pre-clinical experiments. It lowered mHTT mRNA and mHTT protein in a dose-dependent, sustained fashion. Tominersen reversed the HD phenotype, improved survival and reduced brain atrophy in mice. 127 These experiments informed the following phase 1b/2a clinical trial design.
In the initial randomized, double blinded phase 1b/2a clinical trial (NCT02519036), participants (n = 46) were divided into 5 groups of ascending doses and a placebo group. Tominersen was given every 28 days in 4 doses, and participants were followed up after 4 months. 128 No serious adverse events occurred, and minor adverse event incidence was similar for Tominersen and placebo groups. CSF mHTT was reduced by 40% on average at 90 mg and 120 mg doses and Tominersen showed dose dependency.129,130
Multiple trials have commenced since NCT02519036 began. A randomized phase 2 open label extension (OLE) trial (NCT03342053) further assessed adverse events, recruiting people (n = 46) that had participated in the NCT02519036 trial.131,132 Participants were given monthly or bimonthly Tominersen 120 mg doses for 15 months. 129 The trial concluded in 2019, with results yet to be published.
GENERATION HD1 (NCT03761849) is a randomized, double blinded phase 3 trial to measure HD phenotype changes using the Composite Unified Huntington’s Disease Rating Scale (cUHDRS) and total functional capacity (UHDRS-TFC), among other measurements. Researchers will divide participants (n = 909) with manifest HD into 3 groups – placebo, and Tominersen doses (120 mg) every 8 or 16 weeks. Dosing was planned to occur for 25 months, and follow up would have been 29 months.28,133 GENERATION HD1 was expected to conclude in 2022, 28 however, was suspended upon independent data review in early 2021 due to Tominersen’s benefit to risk ratio over time. No new safety signals arose during this review. 29
A randomized phase 3 OLE clinical trial (NCT03842969/GEN-EXTEND) including previous Tominersen trial participants (N = 1100) will measure long-term tolerability, behaviour changes and adverse event incidence.131,134 Participants will receive Tominersen every 8 or 16 weeks, depending on the dose timing of their previous trial. It was expected to conclude in June 2024, 134 but has been placed on hold due to another trial’s benefit to risk ratio. 29
GEN-PEAK (NCT04000594) is a non-randomized, open label phase 1 trial investigating Tominersen’s pharmacokinetics. It will conclude in 2021. Participants (N = 20 in total) in 3 groups of ascending doses will receive 2 doses 28 days apart. CSF Tominersen and mHTT concentrations, and plasma Tominersen concentrations will be measured. 135
WVE-120101 and WVE-120102
Wave Life Sciences’ WVE-120101 and WVE-120102 are allele-specific, stereopure ASOs. They target single nucleotide polymorphisms (SNPs) specific to HD genotypes – WVE-120101 targets rs362307 (SNP1), and WVE-120102 targets rs362331 (SNP2). 136 They are allele-specific, which is thought to potentially avoid long-term negative effects since they do not lower wtHTT. 137 As they target SNPs, WVE-120101 and WVE-120102 cannot treat all HD patients – but combined use could target 80% of European HD patients. 125 Unpublished pre-clinical experiments showed they reduced mHTT mRNA without significantly altering wtHTT mRNA and protein in patient derived cell lines. 138 This led to randomized, double blinded phase 1b/2a clinical trials (PRECISION-HD1 and PRECISION-HD2; NCT03225833, NCT03225846), which recruited participants with early manifest HD and rs362307 (SNP1) or rs362331 (SNP2). Participants (n = 60 per trial) were divided into 6 groups – 2 mg, 4 mg, 8 mg, 16 mg or 32 mg WVE-120101/WVE-120102 doses or placebo (.9% NaCl).126,136 The parallel studies intended to measure adverse events, tolerability, pharmacokinetics and pharmacodynamics. 136
Preliminary PRECISION-HD2 results showed WVE-120102 reduced CSF mHTT protein by 12.4% compared to placebo across all groups (n = 44). This was statistically significant in higher doses, and indicated dose dependency. Total HTT did not change between groups. There were no serious adverse events, and less minor and moderate adverse events for WVE-120102 (72%) than placebo (83%). 126 However, in early 2021 Wave Life Sciences announced PRECISION-HD2 trial data showed WVE-120102 caused no change in mHTT compared to placebo. PRECISION-HD1 data showed similar results, therefore, both trials were stopped. 139
OLE trials began for patients who participated in the PRECISION trials (NCT04617847 for WVE-120101, NCT04617860 for WVE-120102). They received multiple 8 mg or 16 mg doses of WVE-120101/WVE-120102.126,138 OLE results for PRECISION-HD2 indicated slight but inconsistent mHTT lowering, without the ability to consistently lower mHTT with increased doses. Dosing in OLEs has stopped for WVE-120101 and WVE-120102 given their inability to significantly lower mHTT and therefore be of clinical benefit. 139
Wave Life Science’s WVE-003 is a pre-clinical ASO that targets an unspecified SNP (SNP3) in mHTT mRNA. 140 WVE-003 reduced mHTT mRNA in vitro and achieved mHTT mRNA knockdown in vivo. 141 Application for a clinical trial was submitted in late 2020. 140
Other ASOs
BioMarin’s (CUG)7 is a pre-clinical allele-specific ASO which targets expanded CAG repeats in mHTT mRNA. 31 After promising results in HD patient derived fibroblasts, 142 researchers found CUG (7) reduced mHTT protein concentration in R6/2 and Q175 HD model mice by 15-60%, improved motor symptoms and increased cortical volume. 143 R6/2 mice are transgenic HD models created by expressing exon one of the human mHTT gene, containing 150 CAG repeats. 144 Q175 HD knock-in mice express human mHTT within the mouse HD gene, containing 179 CAG repeats. 145
A number of further pre-clinical HD ASO therapeutics are currently in development. Some groups are investigating ASOs that target SNPs associated with HD alleles, therefore, aiming to selectively suppress mHTT expression.146,147 Results in HD mice model neurons, 146 and in vivo using HD model mice 147 were positive. Total HTT reducing and mHTT specific ASOs have improved cognitive and behavioural deficits in HD model mice with safety and tolerability, and reduced HTT in the cortex and limbic structures in non-human primate brains (where cognitive and behavioural symptoms largely originate). 148 Another ASO induces exon 12 skipping in HTT pre-mRNA, preventing cleavage sites being expressed and therefore 586 amino acid N-terminal huntingtin fragments being created. Rodent studies showed successful exon skipping, a shorter HTT protein and reduced 586 amino acid N-terminal huntingtin fragments. 149
Some ASOs are non–HTT-targeting, such as Triplet Therapeutics’ TTX-3360. It targets MSH3 within the DNA Damage Response pathway, which is often involved in repeat expansion. TTX-3360 is safe and tolerable in HD model mice, and 50% MSH3 knockdown reduced or inhibited mHTT repeat expansion. An Investigational New Drug/Clinical Trial Application submission is planned for late 2021. 150
RNA Interference (RNAi) Therapies
RNAi therapies use transgenes that express RNA molecules such as micro-RNA (miRNA), short hairpin RNA (shRNA) and short interfering RNA (siRNA).26,31 Cellular machinery processes these molecules, and they label their target mRNA for degradation. Degradation occurs via enzymes in the RNA-induced silencing complex (RISC), 31 which stops mRNA translation and therefore protein synthesis. 26 RNAi therapies are delivered by a viral vector via intracranial injection. 31 There are advantages and disadvantages associated with RNAi therapies. For example, shRNA therapies can result in difficulty controlling shRNA expression, but shRNA therapies also tend to produce less off target effects than other RNAi therapies and last longer.151-153 These factors, and other factors associated with miRNA and siRNA, must be considered when developing RNAi therapies.
AMT-130
AMT-130 (UniQure Biopharma B.V., named AAV5-miHTT in pre-clinical studies) is a RNAi based gene therapy. It is the only gene therapy currently in clinical trials for HD. 27 AMT-130 contains a gene encoding for a miRNA, which is delivered via adeno-associated virus vector serotype 5 (AAV5). AMT-130 activates RISC, which stops mHTT (and wtHTT) protein synthesis. AMT-130 is administered via intrastriatal injection. 26
In an HD rat model, Hu128/21 HD mice and HD patient derived neurons, AAV5-miHTT reduced mHTT mRNA and protein levels.154-156 Hu128/21 HD mice result from crossing an HD mouse model, YAC128, with a wtHTT model, BAC21, giving them 2 full-length human HTT transcripts that are heterozygous for the HD mutation. 157 Potent mHTT suppression was sustained for 7 months post-injection in Hu128/21 HD mice. 158 In Q175 HD mice, AAV5-miHTT lowered human HTT protein and striatal (39%) and cortical mHTT aggregates 12 months post-injection. R6/2 HD mice showed motor symptom improvement and prolonged survival. 159 mHTT aggregates were suppressed in an HD rat model – reducing neuronal dysfunction. 155 AAV5-miHTT lowered human mHTT mRNA (72.8%) and protein (85.3%) in the minipig brain, and indicated dose dependency. 160 AAV5-miHTT did not create off target effects caused by gene dysregulation or incorrect processing in HD patient derived neurons, 156 but at high doses caused astrogliosis in Hu128/21 mice. 158 AMT-130 showed safety, tolerability and widespread distribution in the brain of non-human primates. 161
Phase 1b/2a clinical trials will test AMT-130’s safety and proof of concept in 26 manifest HD patients (NCT04120493). Some will be given a low or high dose via MRI guided bilateral intrastriatal injection, and others will be given placebo (imitation surgery). 27 Researchers are measuring adverse effects, CSF biomarkers, CSF AMT-130, mHTT concentration and clinical scale changes (like UHDRS) during the core study period and follow up. 162 Data from the initial 6 months of dosing in 2 patients, and 90 day data from the next 2 patients indicated AMT-130 was safe. 163 This prompted complete enrolment of the first dosing cohort (n = 10). The study is expected to be completed in 2026, with an OLE beginning in late 2021. 164
Other RNAi therapies
Voyager Therapeutics’ VY-HTT01 is an allele non-specific RNAi based gene therapy. An miRNA is expressed and delivered via adeno-associated virus vector serotype 1 (AAV1), targeting HTT mRNA degradation. It is administered via intracranial injection. 31 In YAC128 transgenic HD mice, which express full-length human HTT with 128 CAG repeats, VY-HTT01 reduced striatal HTT protein by 40%, partially corrected motor and behavioural symptoms, reduced HTT aggregates and showed effective striatal distribution.165,166 Unpublished results showed VY-HTT01 lowered striatal (67%) and thalamic (73%) HTT mRNA in non-human primates. 167 Clinical trial applications were submitted in late 2020, but are on hold to resolve chemistry, manufacturing and control issues. 168
Spark Therapeutics are also developing an RNAi based drug, using an AAV1 delivered shRNA (AAV.shHD2.1)31. Rodent studies showed motor symptom improvement, reduced human mHTT expression 169 and neuroprotection on administration with AAV.shHD2.1. 170 In non-human primates, lowering HTT expression via RNAi did not lead to neuronal degeneration or an immune response for 6 weeks post-injection into the putamen, strengthening RNAi as a potential HD therapy. 171 Clinical trials have not yet been initiated. 34
RNA Targeting Small Molecule Therapies
Many small molecule therapies alter pre-mRNA to include or exclude target exons, leading to an altered protein (also called splice modulation). They are administered orally, which is advantageous compared to other invasive administration methods. However, this also increases the risk of adverse effects due to their systemic distribution and lack of specificity. 25 All HD RNA targeting small molecule therapies are either pre-clinical or in phase 1 clinical trials currently, with no clinical trial data yet published. 34
PTC Therapeutics’ PTC518 is a RNA splice modulator that induces a psuedoexon inclusion containing a premature stop codon. This enhances mHTT mRNA degradation – resulting in lowered mHTT protein. Unpublished pre-clinical data showed PTC518 is systemically distributed (including widespread central nervous system distribution) and indicated dose dependency. In HD patients’ isolated cells PTC518 lowered mHTT mRNA and protein, and in transgenic BACHD HD mice, which express the full-length human mHTT with 97 mixed CAA-CAG repeats within exon 1, PTC518 systemically lowered HTT protein.172-174 A phase 1 trial has commenced with healthy participants, using ascending single and multiple doses. It is expected to indicate correct dosing, proof of concept and safety. Proof of concept was shown in healthy volunteers with dose-dependent HTT mRNA splicing. It was also well tolerated and showed predictable pharmacology.173,175
Novartis Pharmaceuticals’ Branaplam (also called LMI070) is an oral, weekly administered small molecule RNA splice modulator. It was initially designed to treat spinal muscular atrophy (SMA), 176 but researchers found it lowered mHTT protein in SMA and HD animal models. It also lowered HTT mRNA in HD models. A phase 2b clinical trial with manifest HD participants is planned for in 2021. 177
Nuredis are developing an RNA splice modulator that specifically targets transcription elongation cofactors such as Spt4.34,178 These are required for expanded CAG repeat transcription. Therefore, Spt4 inhibition would result in mHTT protein reduction. Pre-clinical data showed Spt4 inhibition using small molecules reduced expanded polyQ proteins in neurons, while restoring neuronal function and regional enzymatic activity. SUPT4H (a mammalian Spt4 ortholog) also reduced mHTT protein, toxicity and mHTT aggregates in neurons. 178 In R6/2 HD mice, SUPT4H reduction prolonged survival, reduced motor symptoms and reduced mHTT aggregation. Further studies conducted in zQ175 HD mice showed SUPT4H reduced mHTT mRNA and protein. 179
There are other groups that are searching for and developing RNA targeting small molecules to treat HD – including molecules modulating protein disulfide isomerase in the endoplasmic reticulum to reduce mHTT toxicity, 180 molecules altering degradation or autophagy mechanisms 181 and molecules targeting expanded CAG repeat RNA to reduce mHTT protein synthesis.182-184 CAG lengths can be somatically unstable in different tissue types. The mechanism underpinning this is unknown. Understanding and targeting this somatic instability may be a pathway to future therapies. 185
Zinc-Finger Protein (ZFP) Therapies
DNA targeting therapies affect the mHTT gene itself by introducing protein coding sequences to the brain parenchyma that bind to DNA regions. They require a viral vector for delivery and are administered intracranially. 31 ZFPs are synthetically made sequences that mimic naturally occurring motifs. They contain DNA binding components that often include multiple zinc finger peptides, which can bind to 3 to 5 target DNA nucleotides. The major ZFPs currently being developed for HD bind to expanded CAG repeats, preventing the mHTT gene’s transcription. This lowers mHTT protein levels without altering the gene itself. 25 However, zinc finger nucleases (ZFNs) can alter genetic material, allowing for the mHTT gene to be corrected or disrupted. 34 ZFPs have successfully reduced mHTT protein by 60% in the R6/2 HD mice model brain, reduced aggregation and improved motor symptoms. 186
There are 2 major ZFPs in development for HD – TAK-686 and ZF-KOX1.186-188 These therapies are both in pre-clinical stages currently. 25 TAK-686, which targets expanded CAG repeats, has shown success in reducing mHTT in knock-in HdhQ50 mice containing an HD allele with 50 CAG repeats. It also reduced mHTT by 62% and improved behavioural symptoms in R6/2 HD mice.188,189 A modified version of ZF-KOX1 (for mouse compatibility) delayed HD symptoms and inhibited mHTT expression by 77% in HD model mice. 187
Transcription Activator-like Effector Nuclease (TALEN) Therapies
TALENS have DNA binding domains that contain repeating peptides which bind to DNA nucleotides. 34 They cause double strand breaks via artificial nucleases, allowing for segment deletion or correction. 190 All TALENs in development for HD therapy are in pre-clinical or drug discovery stages. Their impacts on phenotype, toxicity and an effective delivery system have not been established yet. TALENs also have not been investigated in HD animal models. 191 However, TALENs have successfully removed expanded CAG repeats in yeast cells by inducing double strand breaks, resulting in gene conversion. They did not increase mutation rate or cause genomic rearrangement. 192
Lowered mHTT gene expression and protein aggregation was also achieved in HD patient derived fibroblasts using a TALEN and SNP specific transcription activator-like effector (TALE-SNP). The TALE-SNP prevented transcription of the mHTT gene by targeting SNPs associated with it, leaving wtHTT gene expression unaltered. TALENs were used for gene correction by creating double strand breaks which were repaired – deleting the expanded CAG repeats. Results showed that some of the TALE-SNPs reduced mHTT expression by up to 20% without altering normal HTT expression, indicating proof of concept.25,191
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-Associated System (Cas) Therapies
Cas9 (CRISPR associated protein 9) is a naturally occurring immune enzyme found in some bacteria, which has been utilized as a genome editing tool for the past decade. It functions via a single-guide RNA (sgRNA) binding to its target DNA, and Cas9 nucleases inducing double strand breaks. 34 Double strand breaks are repaired by error-prone non-homologous end joining (NHEJ), causing frameshifts which can impair gene expression.190,193 All CRISPR approaches are delivered with viral vectors and intracranial injection. 34 In HD therapeutics, CRISPR/Cas9 therapies may remove expanded CAG repeats to correct HD alleles, inactivate HD-associated alleles or target the HTT gene itself, non-specifically reducing HTT. Multiple groups are investigating CRISPR pre-clinically for HD.193-198
CRISPR/Cas9’s efficiency has been assessed in cell lines. A paired Cas9 Nickase strategy successfully excised expanded CAG repeats in exon 1 of the HTT gene in 3 distinct HD patient derived fibroblast lines, inactivating HTT. Nickases cause single strand breaks, reducing off target effects due to increased specificity. Each cell line treated with Cas9 Nickases had different CAG repeat lengths and showed approximately 70% reduction in HTT protein. 193 A strategy using CRISPR-Cas9 and piggyBac transposon was also successful in HD human induced pluripotent stem cells, correcting the mHTT allele and amending HD mediated phenotypic defects. 194
A PAM-altering, mHTT associated SNP based CRISPR/Cas9 approach is also in development. This therapy induces a deletion mutation that inactivates the mHTT allele, leaving an intact wtHTT allele. In HD patient derived cell lines, excision of CAG repeats associated with mHTT alleles was successful. 195
Some groups have investigated CRISPR in HD animal models. Deletion of the mHTT polyQ domain using an allele non-specific CRISPR/Cas9 based strategy successfully repressed human mHTT expression in HD140Q-knock-in mice’s striatum, which express full-length human mHTT with exon 1 containing 140 CAG repeats. CRISPR was administered via striatal injection. This approach reduced mHTT expression, mHTT aggregates, astrocyte reactivity and improved motor symptoms. 196 Another group injected SpCas9 or sgRNAs delivered by recombinant AAVs (abbreviated to rAAV.SpCas9 and rAAV.sgHD1/i3, respectively) into the right hemisphere of BACHD mice. Human HTT exon 1 was deleted, reducing human mHTT mRNA by 40%. The left hemisphere was used as a control, and did not demonstrate lowered mHTT expression. 198 Another group investigated in vivo striatal injection of CRISPR/Cas9 therapies (AAV1-SaCas9-HTT to target human HTT, or AAV1-SaCas9-mRosa26 as a control) in R6/2 HD mice. They found AAV1-SaCas9-HTT had reduced mHTT protein inclusions by 40%, reduced total mHTT protein by 50%, and it improved lifespan and motor symptoms. 197
Stem Cell Therapies
Stem cell therapies are another candidate for future HD therapeutics (Figure 3). Their main advantages are their potential ability to replace neurons lost due to HD pathology, improve regeneration and provide pro-survival factors. Furthermore, theoretically some long-term benefits would remain due to stem cell incorporation into the patient’s brain microenvironment. However, there are also concerns these therapies may induce immune reactions or rejection.
199
Cellavita HD, foetal striatal stem CRT and autologous stromal/adipose-derived stem cell therapy development timeline.
Cellavita HD
Cellavita HD is the most developed stem cell therapy for HD currently 27 (Figure 3). It is a mesenchymal stem cell therapy, and is administered via intravenous injection. 200
Azidus Brasil began a non-randomized, open label phase 1 clinical trial in 2017 to test safety and effectiveness of their stem cell therapy for HD, with completion expected in 2023 (NCT02728115/SAVE-DH).27,200 Participants (n = 6 total) with manifest HD will be given 3 low or high monthly Cellavita HD doses. They will be followed up for 5 years to track adverse events and efficacy through the UHDRS scale and inflammatory markers, among other measurements. 200
A randomized, triple blind phase 2 clinical trial began in 2018 to test Cellavita HD’s dose-response relationship (NCT03252535/ADORE-DH).27,201 Thirty-five participants with manifest HD will be given 9 total low or high doses of Cellavita HD or placebo. Researchers are primarily measuring UHDRS to assess effective dosing. This trial will conclude in 2022. 201
An OLE phase 2/3 trial has also been registered, but is not yet recruiting (NCT04219241/ADORE-EXT). It will assess longer term safety and efficacy in 35 manifest HD participants who took part in the ADORE-DH trial.27,202 They will receive multiple doses over 24 months. UHDRS, TFC, neurological image improvement and clinical worsening will be measured, among other factors. It will conclude in 2022. 202
Foetal striatal stem cell replacement therapy
Foetal striatal stem cell replacement therapy was first investigated in HD patients over 20 years ago. Multiple early studies showed foetal striatal cell replacement therapy (CRT) was safe but relatively ineffective at altering patient symptoms. A phase 1, randomized controlled trial using unilateral intrastriatal grafts in 4 HD patients indicated no adverse events but no HD symptom improvement (ISRCTN36485475). 203 Similarly, a later trial using bilateral foetal striatal CRT grafts showed the UHDRS score was similar across those with (n = 5) and without (n = 12) striatal grafts. The grafts did not cause significant adverse events. 204
Some trials have shown HD symptom improvements. In 5 HD patients, asynchronous bilateral intrastriatal transplants of foetal striatal neuroblasts created increased striatal metabolic activity, cognitive and motor improvements in 3 of the 5 patients (compared to 22 control HD patients). However, the other 2 patients’ condition declined similarly to the control group. 205 Another group also found mild short-term symptom improvement in 6 patients, but this only persisted in 3 and did not alter overall HD progression. 206
A randomized, open label, phase 1 clinical trial using a relatively higher CRT dose to previous studies was initiated in 2017. CRT will be administered to 5 HD patients via intrastriatal injection and patients will receive immunosuppressant medication (ISRCTN52651778/TRIDENT).27,207 TRIDENT aims to assess safety at 4 weeks, and the effectiveness of an increased CRT dose. The trial will conclude in 2023. 207
However, foetal striatal CRT also raises concerns. Immunosuppressants used to reduce graft rejection have been problematic due to side effects and lowered compliance. 208 Immune responses can occur due to foetal striatal stem cell grafts – over a decade after the ISRCTN36485475 trial, 1 of the 4 patients showed strong microglial activation, immune cell infiltration and mHTT protein aggregates at the graft site. 209 However, while some groups believe immunosuppressants are required, 210 others have shown patients’ immune responses after CRT grafts are unpredictable – therefore excessive immunosuppressant therapy is undesirable. 211 Some groups have found that striatal foetal neural grafts can only provide clinical improvement for a few years, after which patients decline. Therefore, CRT may not be viable as a permanent cure. 212 Ethical concerns also persist due to the use of foetal cells, and graft survival is uncertain due to findings showing reduced vascularization, and lowered astrocytes and pericyte numbers. 213
Autologous stem cell therapies
Autologous stromal cells are also being investigated by Regeneris Medical to treat a range of neurological disorders in a current clinical trial (n = 300), including HD (NCT03297177). 27 Autologous stem cell transplantation requires reprogramming mutated cells or isolating cells with the wild type allele. 214 Pre-clinical data for autologous adipose derived stem cells transplanted bilaterally into the striatum were positive in R6/2 HD model mice. This approach reduced striatal apoptosis, prolonged lifespan, reduced mHTT aggregates and modulated striatal neuronal loss. 215
A non-randomized, placebo-controlled study will assess the safety and change in neurological function of those given autologous stem/stromal cells compared to normal saline for up to 5 years. 27 Autologous cells will be collected from patients’ subdermal fat, isolated, concentrated and neutralized before being given intravenously. The study is expected to be completed in 2023. 216
Mesenchymal stem cell therapies
Mesenchymal stem cells (MSCs) have also been trialled in HD animal models. MSCs originate in the umbilical cord (UC), amniotic fluid, bone marrow and adipose tissue. They are self-renewing, can release neuroprotective factors like BDNF and can differentiate into multiple cell types, potentially including neural cells.217,218 No teratomas or overgrowths have been found in MSC animal model studies. 217 Striatal injection of mice UC-derived MSCs in R6/2 HD mice improved spatial memory and reduced neuropathological deficits, but did not change motor impairments. 219 Striatal transplantation of UC-derived rat MSCs in 3-nitropropionic acid (3-NP) lesioned HD rats improved motor coordination, reduced striatal atrophy, prevented oxidative stress in PC12 cells and enhanced cell viability. 220 3-NP HD rats were systemically, subcutaneously administered with 3-NP, a mitochondrial toxin, to produce striatal lesions. 221 Another 3-NP HD rat study found injection of bone marrow derived rat MSCs resulted in no significant striatal inflammation, no lateral ventricle enlargement, reduced neurological and behavioural deficits and the presence of striatal neurotrophic factors. 222
Induced pluripotent stem cell-derived neural stem cells
Induced pluripotent stem cell-derived neural stem cells (iPS-NSCs) are self-renewing, reprogrammed somatic adult cells that can differentiate into many cell types. 223 Striatal injection of mice iPS-NSCs in an HD rat model was trialled and monitored using serial 18F-FDG PET/CT. Researchers found that the rats’ striatal volume was partially recovered, memory, learning and glucose metabolism improved and iPS-NSCs differentiated into striatal neurons and glia. 224 Quinolinic acid HD rats undergo striatal injections of quinolinic acid to induce an HD phenotype. 225 Another study in a quinolinic acid-induced HD rat model found striatal transplantation with human iPS-NSCs reduced behavioural deficits, reduced inflammation, promoted neurogenesis and replaced some neural cells. 226 Striatal injection with control mice iPS-NSCs into YAC128 HD mice resulted in motor improvement, increased striatal BDNF and iPS-NSC differentiation into neural cells. 227
Antibody Therapies
Antibody-based therapies are not only being evaluated for a range of tauopathies and synucleinopathies, but are also emerging as a potential therapy for monogenic disorders of the central nervous system, including HD (Figure 4). ANX005 and VX15/2503 antibody therapy development timeline.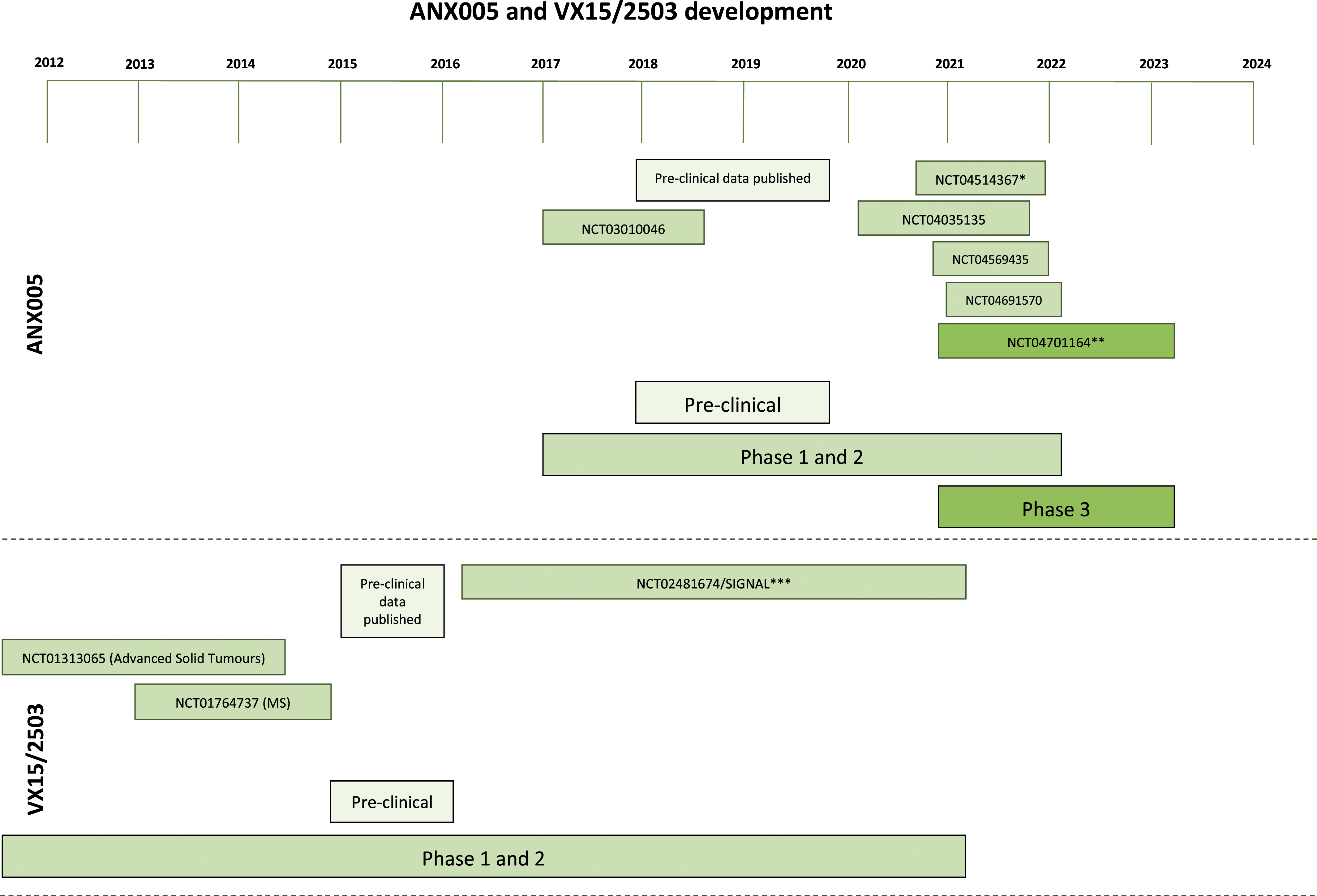
ANX005
Annexon, Inc’s ANX005 is a monoclonal antibody that targets C1q, preventing classical complement cascade initiation. 228 When neuronal stress is already implicated during HD pathogenesis, C1q′s role in synaptic pruning can be disrupted – affecting synapse loss, neurodegeneration and neuroinflammation. Through C1q inhibition, ANX005 is thought to reduce synapse loss and neurodegeneration.229,230 ANX005 was initially intended to treat Guillain-Barré Syndrome (GBS) and Alzheimer’s Disease (AD). 228
Pre-clinical studies showed high affinity binding of ANX005 to C1q in multiple species, and successful CSF and serum C1q reduction in monkeys and rats. Complement cascade inhibition in vivo and in vitro in GBS and AD mice models was successful. Safe dosing frequency, dose dependency and tolerability was also established in monkeys and rats. 228 A murine version of ANX005 reduced neurofilament light chain and synapse loss in HD animal models. 229
A randomized, placebo-controlled, double blinded phase 1 clinical trial began in late 2016 in 27 healthy volunteers via an ascending dose schedule (NCT03010046). ANX005 was given intravenously, either on its own or with intravenous immunoglobulin (IVIg). The study aimed to assess safety, pharmacokinetics and pharmacodynamics. It was terminated in 2018 since there were no adverse events at the maximum dose. 231 Subsequently, investigators began testing ANX005 for HD, GBS, Amyotrophic Lateral Sclerosis (ALS) and Warm Autoimmune Haemolytic Anaemia (wAIHA) patients.232-235
A phase 1b clinical trial in 31 GBS patients showed promising top-line results using an ascending dose schedule. Its aims were to test safety, tolerability and pharmacokinetics. The ANX005 dose was increased after safety was established in up to 100 mg/kg in 19 participants. No significant adverse events occurred, and ANX005 successfully inhibited C1q. ANX005 was associated with early muscle strength improvement, reduced neurofilament light chain and small GBS disability scale improvements. 236 ANX005 is also in a phase 1b, open label clinical trial to assess safety in 12 GBS patients (NCT04035135), 232 and a randomized, double blinded phase 2/3 trial in 180 GBS patients to assess efficacy and safety (NCT04701164). 237
There are also 2 phase 2, open label studies in progress for ALS and wAIHA (NCT04569435, NCT04691570, respectively). ANX005 will be given to 24 participants with ALS for 12 weeks (NCT04569435), and given to 12 patients with wAIHA for 2 doses (NCT04691570). Both studies will test ANX005’s safety, tolerability, pharmacodynamics and pharmacokinetics. The ALS trial will end in late 2021, and the wAIHA trial in early 2022.234,235
An open label phase 2a clinical trial will evaluate ANX005’s safety, tolerability, pharmacokinetics and pharmacodynamics in HD patients or patients at risk for HD (n = 24) (NCT04514367). Seven ANX005 infusions will be given for up to 10 weeks, then patients will be followed for 3 months. The study will be completed in late 2021. 233
VX15/2503
Vaccinex’s VX15/2503 (also called Pepinemab) is an IgG4 monoclonal antibody that inhibits protein semaphorin 4D (SEMA4D). SEMA4D is a protein associated with neuroinflammation by promoting glial cell activation and activating signalling cascades causing oligodendrocyte and neural precursor cell apoptosis, among other processes. 238
Pre-clinical studies showed that VX15/2503 did not induce toxicity or adverse events in rats or non-human primates. It lowered T-cell SEMA4D and induced long-term saturation with 5 weekly doses. Dose dependency was established. 238 Anti-SEMA4D is not immunosuppressive and improves blood brain barrier integrity. 239 Another study found that anti-SEMA4D improved cortical and striatal atrophy, and cognitive symptoms in YAC128 HD model mice. It did not influence motor symptoms. 240
Vaccinex, Inc and the Huntington Study Group conducted a double blinded, phase 2 clinical trial testing VX15/2503’s safety, tolerability and pharmacokinetics in late prodromal and early manifest HD patients (NCT02481674/SIGNAL). Patients were divided into 2 cohorts, A (12 months treatment) and B (36 months treatment), and given intravenous monthly VX15/2503 doses or a placebo. Investigators assessed MRI brain volume, motor and cognitive function and CSF biomarkers, among other measures. The study ended in mid-2020.241,242
Preliminary SIGNAL results from Cohort A (n = 36) showed VX15/2503 was tolerable and safe. Reduced metabolic activity and brain volume reduction appeared to be prevented. Cohort A were given VX15/2503 for another 5 months (open label), with a 3 month follow up. 243 However, results from cohort B1 with early manifest HD (n = 179) found that VX15/2503 did not prevent brain volume and metabolic activity reduction, or significantly alter motor, cognitive or behavioural function. 244 Trends towards cognitive benefits were found, and post-hoc analysis using Clinical Global Impression of Change in an advanced HD subpopulation confirmed this. However, these findings require further investigation. 245
VX15/2503 is also in clinical trials for AD, head and neck cancer and colorectal cancer,246-249 and has been tested in Advanced Solid Tumours, Multiple Sclerosis (MS), melanoma, non-small cell lung cancer.250-252 Advanced Solid Tumour (NCT01313065) (N = 42) and MS (NCT01764737) (N = 10) trials found VX15/2503 was safe and tolerable.250,251
Anti-mHTT antibody therapies
Anti-mHTT antibodies have been tested pre-clinically for over 20 years. Their intracellular targets, such as the PolyQ, PolyP and N-Terminal exon 1 domains, are affected using intrabodies. 32 Intrabodies are simpler forms of antibodies delivered via viral vectors that are able to block and inactivate proteins, enhance degradation and stop protein misfolding intracellularly. 253
PolyQ targeting intrabodies have had negative results, but PolyP/proline-rich region and N-Terminal exon 1 domain targeting intrabodies have had mixed results. rAAV6-INT41 (developed by Vybion Inc) is a PolyP/proline-rich region targeting intrabody that can lower gene dysregulation caused by mHTT.32,254 It was delivered in R6/2 HD mice by a viral vector (rAAV6) via bilateral intrastriatal injection. INT41 can reduce small mHTT aggregates by 31% and larger aggregates by 16%, while improving cognitive deficits in R6/2 HD mice. 254
Intrabodies targeting mHTT aggregates have had positive results, but raise concerns since conformational antibodies can also increase aggregation.
32
W20 is an oligomer specific intrabody that targets mHTT aggregates, among other aggregates, such as
Other intrabodies have also been tested pre-clinically, such as HAPP1 and VL12.3. These target the P-rich epitope of the mHTT protein, and the N-terminal 17 amino acids, respectively. They were found to lower HTT exon 1 toxicity and aggregation. 256 C4 also targets the N-Terminal of HTT fragments. 257 When fused with mouse orthenine decarboxylase including a deleted C-terminal PEST motif (mODC-PEST), HD exon 1 fragments are reduced, along with lowered HD exon 1 fragment toxicity, aggregation and intrabody degradation. 258
Arguably antibody therapies’ main advantage is their ability to target extracellular mHTT, since most other therapies cannot do so. This is achieved via active (self-produced antibodies) or passive (pre-made antibodies) immunization. 259 Active immunization’s functional benefits are not yet known, and raise safety concerns due to increased immune gene dysregulation. Passive immunization has decreased mHTT in YAC128 mice. 32 Furthermore, AFFiRiS′ mAB C6-17 effectively bound and demonstrated specificity to human mHTT in cell-based assays and immunohistochemistry. C6-17 targets a site within the HTT protein near the aa586 caspase-6 cleavage region, and has blocked extracellular mHTT uptake. Distribution of C6-17 was quick and widespread in HD model mice, showed reduced mHTT in the central nervous system and improved motor symptoms.260,261
Antibody therapies may be most beneficial in combination with RNA based therapies to target both intracellular and extracellular mHTT. 259
Other Small Molecule Therapies
Pridopidine
Pridopidine (also named TV-7820; formerly named ACR16 or Huntexil) is an oral small molecule developed by Prilenia Therapeutics (previously developed by Teva Pharmaceutical Industries Ltd) for HD motor symptoms
262
(Figure 5). It is a dopamine stabilizer that binds to dopamine type 2 receptors, therefore, affecting striatal dopamine pathways involved in motor HD pathophysiology.262,263 It also activates the sigma-1 receptor, increasing BDNF production and supporting neuroprotection.262,264 Pre-clinical studies in R6/2 HD mice showed pridopidine prevented apoptosis, increased pro-survival molecules and reduced mHTT aggregate size – indicating neuroprotection. It also improved motor function.
265
Pridopidine prevented medium spiny neuron loss in YAC128 HD mice.
266
Non-RNA targeting small molecule (Pridopidine, Laquinimod, VX-745 and Nilotinib) therapy development timeline.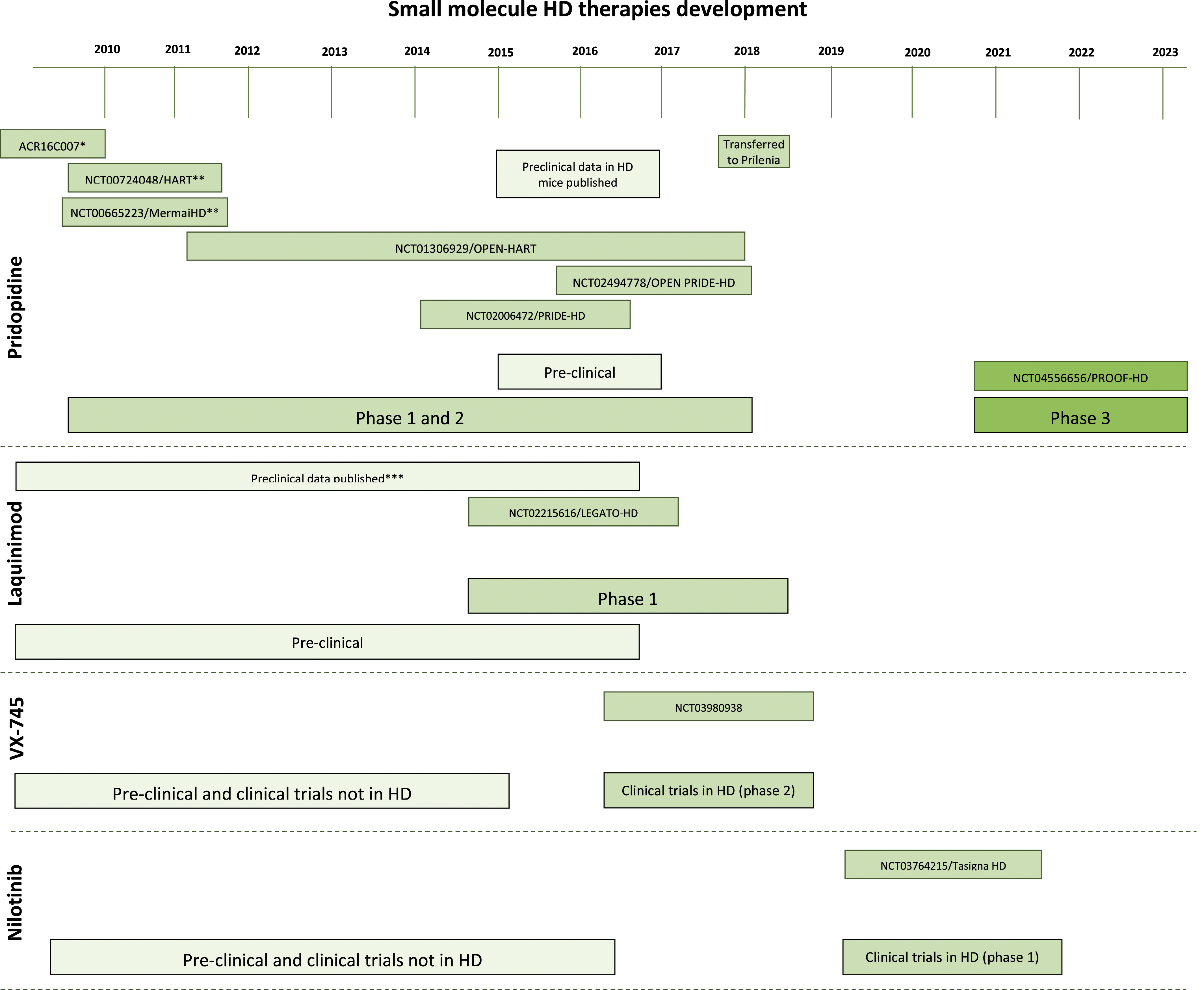
Pridopidine was first clinically investigated as an HD therapy in 58 HD patients in a randomized, double blinded trial (ACR16C007). It showed tolerability and safety. While cognitive scores did not differ between placebo and pridopidine, voluntary motor performance improved using pridopidine. 267
Further trials assessed efficacy and safety of pridopidine. In a 26-week, double blinded phase 3 clinical trial (NCT00665223/MermaiHD), 437 HD patients were randomized to 45 mg or 90 mg pridopidine, or placebo. Modest motor improvements were found using UHDRS-total motor score (TMS), but this was a tertiary endpoint and other improvements (like the mMS) were not statistically significant. 268
A 12-week double blinded phase 2/3 clinical trial (NCT00724048/HART) randomized 227 HD patients to 3 dose groups or placebo. No statistically significant modified motor score (mMS) improvements were found, but trends pointed towards voluntary motor function improvement at 90 mg. 269 Both studies found acceptable tolerability and safety. Similarly, a 26-week, double blinded, phase 2 clinical trial found tolerability (NCT02006472/PRIDE-HD). 408 HD patients were randomized to 4 differing pridopidine dose groups or placebo. Investigators found no UHDRS change between placebo and pridopidine groups. A few serious adverse events occurred that likely related to HD symptoms, and 1 death during the trial was possibly affected by pridopidine administration.263,270
The PRIDE-HD trial was extended from 26 to 52 weeks in 262 patients. At 52 weeks, those taking 45 mg of Pridopidine had a lowered TFC compared to placebo, indicating slowed HD progression. Patients administered 45 mg did not experience TFC decline for the entire year they were taking pridopidine. They also found Pridopidine also has a dose-specific effect. 271
These findings led to an OLE for the MermaiHD trial and for the HART trial (NCT01306929/OPEN-HART). Both studies found long-term tolerability and safety.272,273 In OPEN-HART, 225 HD patients were administered 45 mg Pridopidine twice daily over 36 months. The trial found HD progressed similarly between placebo and Pridopidine groups for the first 36 months (as measured by TFC and TMS). 273 However, additional data showed that while TFC declined over the full 60 months, the rate of decline slowed for those taking pridopidine compared to placebo. TFC and TMS also remained stable between 48 and 60 months. However, the sample size had dropped to 33 patients by 60 months which could create confounding due to those remaining in the trial potentially having a less aggressive disease form. 274 An OLE study was also initiated for PRIDE-HD participants (NCT02494778/OPEN PRIDE-HD) 275
A double blinded, phase 3 clinical trial (NCT04556656/PROOF-HD) will investigate efficacy (via UHDRS-TFC score) and safety in 480 HD patients. Participants will be randomized to 45 mg Pridopidine twice daily or a placebo. The study will end in 2023 and an OLE is planned for PROOF-HD participants. 276
Laquinimod
Laquinimod, developed by Teva Pharmaceutical Industries, is an orally administered immunomodulator therapy
277
(Figure 5). It likely reduces central nervous system leukocyte infiltration and is neuroprotective by stimulating BDNF production, but its precise mechanism of action is unclear.
278
Pre-clinical studies showed laquinimod decreases microglial activation, reactive astrogliosis, astrocytic NF-
Laquinimod was tested in a randomized, double blinded, 52-week phase 2 clinical trial assessing efficacy (via UHDRS-TMS and other clinical measurements), safety and caudate volume atrophy (NCT02215616/LEGATO-HD). Participants (N = 352 total) with HD received 0.5 mg, 1 mg or 1.5 mg laquinimod or placebo.277,282 The 1.5 mg dose was terminated in 2016 due to safety concerns prompted by a study on laquinimod for MS. 277 LEGATO-HD did not show a statistically significant improvement in UHDRS-TMS, but caudate volume significantly improved. Laquinimod was safe and well tolerated. 283 However, further analysis showed that astrocytosis and gliosis was reduced in laquinimod groups. 284 Some Q-motor measurements were also statistically significantly improved in 0.5 mg laquinimod, however, multiplicity corrections were not performed. 285
VX-745
EIP Pharma’s VX-745 (also called Neflamapimod) modulates neuroinflammation and microglial dysfunction, and decreases microglial proinflammatory cytokine upregulation via p38α MAPK inhibition.286,287 VX-745 has been investigated for over 20 years,
288
but has only recently been trialled for HD
27
(Figure 5). In AD model mice, VX-745 reduced hippocampal interleukin-1 beta (IL-1
EIP Pharma Inc and Voisin Consulting, Inc will fund a randomized, placebo-controlled, double blinded, 10-week phase 2 clinical trial in patients with early HD using orally administered VX-745 (n = 16) (NCT03980938). 27 It will primarily assess hippocampal dysfunction changes after 20 weeks. The study began in 2019 and is currently recruiting. 289
VX-745 treatment has also been assessed in AD and Lewy Body Dementia (LBD). A trial in LBD showed cognition and mobility benefits with VX-745 administration, 290 but a study on mild AD did not indicate statistically significant clinical benefit. 291 All studies to date have shown VX-745 is safe and tolerable.290,291
Nilotinib
Nilotinib (also called Tasigna® and AMN107) is being investigated as an oral HD therapeutic27,292 (Figure 5). It is a BCR-ABL tyrosine kinase inhibitor that traditionally treats chronic myeloid leukaemia (CML). 292 Nilotinib reduced oxidative stress in Parkinson’s Disease (PD) model mice via ABL inhibition and improved motor performance. 293 Tyrosine kinase inhibitors also modulated misfolded protein pathophysiology by autophagic clearance in PD model mice, positively affecting neuroinflammation. 294 Clinical trials with PD and LBD participants (n = 12) used lower Nilotinib doses than those that treat CML. Investigators found Nilotinib was safe and tolerable, and appeared to inhibit central nervous system ABL. However, the study was small and not placebo-controlled, necessitating further trials. 292 Georgetown University are recruiting for an open label phase 1 clinical trial to assess safety, tolerability and CSF biomarkers associated with motor and behavioural HD symptoms (n = 20) (NCT03764215/Tasigna-HD).27,295 Ten participants with HD will be given 150 mg for 3 months, and if safe and tolerable another ten participants will be given 300 mg for a further 3 months. There is no control group. 295
PF-02545920
Multiple therapies have been discontinued despite promising pre-clinical results. Pfizer’s PF-02545920 is an oral phosphodiesterase 10a (PDE10a) inhibitor.277,296 It aims to treat HD motor symptoms. PDE10a inhibition improves basal ganglia pathology associated with HD, such as improving spiny projection neuron pathology and responsiveness in HD mice. 297 A randomized, double blinded, 26-week phase 2 trial assessing safety and efficacy (via UHDRS-TMS) of PF-02545920 compared 5 mg and 20 mg PF-02545920 with placebo in 272 HD patients with chorea (NCT02197130/Amaryllis).277,296 Results showed no change in UHDRS-TMS or secondary endpoints between placebo and PF-02545920, despite PF-02545920 being safe and tolerable. An OLE study for Amaryllis participants was terminated after these results, 277 and PF-02545920 was discontinued from Pfizer’s drug development pipeline. 298
Other small molecule therapies are also being investigated. Fenofibrate is a peroxisome proliferator-activated receptor (PPAR) agonist, 27 capable of modifying γ coactivator-1α (PGC-1α). 299 PGC-1α is a transcriptional co-regulator involved in mitochondrial malfunction in HD, leading to neuronal degeneration. 300 Fenofibrate is in a 6 month, randomized, placebo-controlled, double blinded, phase 2 clinical trial assessing safety and efficacy (NCT03515213). 27 Azevan Pharmaceuticals’ SRX246 is a vasopressin 1a receptor antagonist that has completed Phase 2 clinical trials (NCT02507284), aimed at treating HD patients with irritability and aggression. It showed safety, tolerability and validity as a treatment candidate in the near future. 301 Varenicline is a nicotinic acetylcholine receptor agonist targeting cognitive improvements in early HD patients who smoke. HD patients treated with varenicline showed significant executive function and emotional recognition improvements, without clinically significant adverse events. 302 Sage Therapeutics’ SAGE-718 is an NMDA receptor positive allosteric modulator (PAM) that targets HD cognitive symptoms. Phase 1 results showed tolerability, no clinically significant adverse events and improved cognitive performance from baseline, leading to planned phase 2 trial initiation for late 2021.303,304 PBT2 likely reduces metal induced mHTT aggregation through metal protein attenuation. It has completed phase 2 trials (NCT01590888), showing tolerability and safety but no clear benefit for cognitive scores – warranting further studies. 305
Further Neuroinflammation Targeted Therapies for HD
Minocycline
The presence of chronic inflammation in HD has presented a potential therapeutic target with a number of treatments targeting inflammation being proposed. The second-generation tetracycline drug, minocycline, was 1 such proposed therapy that showed anti-inflammatory and anti-apoptotic properties. Mice treated with a low dose of minocycline brought about better behavioural performance than HD mice without treatment (5 mg/kg/day). 306 Stabilization of general neuropsychological and motor symptoms along with significant improvements in psychiatric symptoms were seen in patients administered with 100 mg/day of minocycline in a 2 day clinical trial. 307 However, a low sample size of 11 patients suggests that these results must be corroborated with similar studies on a larger number of patients. A further clinical trial of minocycline treatment on 30 patients over a 6 month period reported a trend toward symptom improvement on the URHDS. 308 The lack of statistical significance in these results were attributed to the short overall treatment period with the study suggesting the need for a period longer than 6 months for effective minocycline treatment. A larger study of 87 HD patients over 18 months assessed the potential benefits of this drug treatment in a phase 3 clinical trial (clinicaltrials.gov ID: NCT00277355) and found that patient improvement did not reach a futility threshold. 309 Based on these findings, further trials of minocycline administration at 200 mg/day were not warranted. 309
Cannabidiol
Cannabinoids have also been postulated to be potential therapeutic targets due to their anti-inflammatory properties.310-312 A clinical trial of cannabidiol treatment on 15 HD patients showed no improvement in motor function over a 6-week period. 313 The low sample size of this study again poses an issue when concluding the effectiveness of cannabidiol as a therapeutic agent. However, a double-blind, randomized, placebo-controlled, cross-over pilot clinical trial with Sativex (®), a botanical extract with an equimolecular combination of delta-9-tetrahydrocannabinol and cannabidiol, conducted over 12 weeks on 24 HD patients, also reported no significant symptomatic effects (clinicaltrials.gov ID: NCT01502046). 314
The failure of these clinical trials may suggest that global immunosuppression (for example through corticosteroids) or the effects of anti-inflammatory treatments may be too broad and may also highlight a need to consider the heterogeneity of microglial and astrocytic phenotypes.315-317 A coherent understanding of the diverse characteristics of these cells and their functions in a healthy state, different disease states, infection, trauma, development and ageing is critical for better therapeutic outcomes.
Conclusions
Positive steps are being made in the development of current HD therapeutics. Guidance for clinicians on HD treatment is improving – guidelines were released relatively recently to elevate international standards of HD care. 116 Current therapies focus on symptom management via multidisciplinary teams, using pharmacological and non-pharmacological treatment. 318 However, HD management has not significantly advanced for the past 20 years. 52 Furthermore, diverse and changeable symptom presentations creates complexity when treating HD. 18 There is no cure for HD, or therapeutic to alter disease onset or progression. 116 HD management requires a tailored approach not only due to symptom diversity, but also adverse effects, common contraindications, drug-drug interactions and certain HD medications worsening other symptoms.18,52,319 Clinicians must navigate these issues in addition to effectively treating HD symptoms.
Perhaps the most prominent shortcoming of today’s HD therapeutics and previous experimental therapeutics is their inability to impact specific HD targets, instead influencing non-specific and downstream processes. 30 This limits their efficiency as disease mechanisms will still propagate and cause harm where they are not inhibited. Pursuing HD-specific targets at the root cause of disease is therefore the new and likely future research direction. 30 The recent surge in interest to develop therapies that target mHTT RNA and DNA shows this trend. 31 Additional therapeutic targets are being investigated, including dopaminergic and glutamatergic neurotransmission, synaptic activity, BDNF signalling, mitochondrial function and biogenics, and aggregate inhibition.320-330 However, several of these targets were primarily researched over 10 years ago, and do not appear to be areas of significant interest in the field currently. Peripheral targets are also being investigated due to peripheral HD symptoms and HTT expression.331-333 For example, the gut microbiome is a key target due to its bidirectional communication with the brain and alteration in HD.334-338
RNA therapies, particularly antisense oligonucleotides, have favourable results and are currently of the most promising future HD therapeutics.26,339 However, RNA targeting therapies come with their own challenges. It is unknown whether non-selective HTT lowering is safe, if off target effects will occur in larger populations, whether mHTT is the singular cause of HD pathology and if permanent HTT suppression is required. Furthermore, if allele-specific treatments are required, 1 SNP will not treat all HD patients.137,340 Many RNA based therapies also require invasive administration. 31 Whether this is tolerable and effective in the long term for larger populations will likely be determined by future research. Initial clinical findings from RNAi therapies and RNA targeting small molecules, and further clinical results from antisense oligonucleotides will clarify RNA based therapies’ efficacy for HD patients. Particularly, results of larger trials such as PRECISION-HD1 and PRECISION-HD2, and further information on GENERATION-HD1 will likely inform whether antisense oligonucleotides remain the frontrunners of future HD therapeutics.
DNA therapies may also enter clinical trials within the next decade, but much work still needs to be done to achieve this goal. Ensuring DNA therapies, particularly CRISPR therapies, are safe for human use will be of primary importance. 137 Combination therapies may also be explored in future research. Current research suggests combining multiple RNA targeting therapies, 341 or RNA targeting therapies with antibodies or stem cells32,214 may be beneficial.
Work on future HD therapeutics will continue for numerous years to come and resulting treatments for HD patients are likely some time away. 34 However, many of the latest developments are encouraging and a new era of HD therapeutics may be approaching.
Footnotes
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Oakley Mental Health Research Foundation (AK, MF, TP, HJW, RLMF; 3725204), Alzheimers New Zealand Charitable Trust (AK; 370836), Alzheimers New Zealand (AK; 3718869), Freemasons New Zealand (AK; 3719321), Aotearoa Foundation, Centre for Brain Research and University of Auckland (AK; 3705579), Neurological Foundation of New Zealand (AK, TP; 848010), Brain Research New Zealand (AK, HJW, RLF), Health Research Council of New Zealand (RLF and HJW; 3627373).