
Abstract
Background
Recently, the number of available disease modifying therapies for multiple sclerosis (MS) has increased. However, a proportion of patients treated with these agents continue to experience relapses and disease progression. Cladribine tablets, approved in 2017 for highly active relapsing MS, comprise a sparsely administered oral treatment which exerts its therapeutic effect through a reduction and subsequent repletion of the lymphocyte population.
Purpose/Study Sample
Here we describe the design of CLAD CROSS, a prospective, non-interventional, multicenter, Phase IV study in patients with a confirmed diagnosis of RRMS who switch from first-line disease modifying drugs (DMDs) to treatment with cladribine tablets in routine clinical practice. 242 adult patients will be recruited in 61 sites (6 countries) over 30 months and will be followed up for 2 years following prescription of cladribine tablets per the decision of the treating physicians.
Research Design
The primary endpoint is the change in annualized relapse rate (ARR) between the 12-month pre-baseline period and over the 12-month period before end of study. Secondary endpoints are the percentage of patients with 6-month disability progression or improvement at the end of the study, measured by the Expanded Disability Status Scale, Timed 25 Foot Walk and 9-Hole Peg Test scales and quality of life, treatment satisfaction, and healthcare resource utilization, measured through the MSIS-29, TSQM 1.4, and EQ-5D-3L scales, respectively. MRI lesions will be compared in the exploratory setting between the 12-month pre-baseline period, baseline, and at years 1 and 2. Adverse events will be monitored throughout the study. Interim analyses are pre-planned when 30% and 60% of patients will complete the 12-month follow-up visit.
Conclusions
CLAD CROSS will provide efficacy data on cladribine tablets, used as a follow-up treatment to first-line DMDs in the real-world setting, will further establish its safety profile and will collect information to support pharmacoeconomic studies.
Introduction
Recently, the number of available disease modifying therapies for the treatment of relapsing–remitting multiple sclerosis (RRMS) has increased. However, a proportion of patients treated with these agents continue to experience relapses and disease progression. 1 Cladribine is a nucleoside analog of deoxyadenosine. The cladribine prodrug is phosphorylated intracellularly to its active product, 2-chlorodeoxyadenosine triphosphate (Cd-ATP), by deoxycytidine kinase. This deoxynucleotide product is degraded in most cells, by 5′-nucleotidase. Cells such as lymphocytes that contain a high deoxycytidine kinase activity but low 5′-nucleotidase activity, that is, a high deoxycytidine kinase to 5′-nucleotidase activity ratio, accumulate deoxynucleotides to high concentrations, resulting in lymphocyte cell death. By this mechanism, cladribine tablets exhibit a selective mode of action on B and T lymphocytes, resulting in targeted and sustained reductions of these cell populations. 2 In the Phase III CLARITY study, treatment with cladribine tablets given in short courses annually for 2 years significantly improved clinical and MRI outcomes. 3 In the Phase III ORACLE-MS study, cladribine significantly reduced the risk of clinically definite MS in patients with a first clinical demyelinating event compared with placebo. 4 The Phase II ONWARD study has shown a significant reduction in annualized relapse rate (ARR) when comparing the pre-baseline treatment period with the cladribine tablet treatment period. However, ONWARD only allowed IFNs as pre-baseline treatments; it did not include other first-line treatments [DMF (dymethylfumarate) and teriflunomide (TFN)] and it was not powered to detect differences in the ARR. 5
Here we describe the design of a prospective, non-interventional, multicenter, Phase IV study in patients with a confirmed diagnosis of RRMS who switch from first-line disease modifying drugs (DMDs: interferons, glatiramer acetate, TFN, or DMF) to treatment with cladribine tablets in routine clinical practice. The primary objective is to evaluate the change in ARR between the 12-month pre-baseline period and over the 12 months period before end of study follow-up (2 years). Secondary objectives are to study (1) the change in ARR over the 12 months following the switch to cladribine tablets, (2) the percentage of patients with disability progression or improvement at the end of the study compared to baseline, (3) patient satisfaction with the treatment and its impact on quality of life, and (4) to obtain real-world pharmacoeconomic data on the use of cladribine tablets. Exploratory objectives are to compare MRI findings between baseline and years 1 and 2, as well as between the 12-month pre-baseline and post-baseline results (years 1 and 2). A subgroup analysis per pre-baseline DMD will also be conducted.
Methods
Participants, Interventions, and Outcomes
Study Setting
61 sites will be included in the study, in 6 countries (Greece, Italy, Switzerland, Norway, Poland, and Portugal). The list comprises academic neurology departments as well as public and private sector neurology departments. The complete list of sites can be obtained from the Sponsor.
Eligibility Criteria
Inclusion and Exclusion Criteria.

Interventions and Participant timeline
This is a prospective, non-interventional, multicenter, Phase IV study in patients with a confirmed diagnosis of RRMS who switched from first-line DMD treatment (interferon, glatiramer acetate, teriflunomide, and DMF) to treatment with cladribine tablets in routine clinical practice.
No study specific interventions will be performed on the patients and there will not be any study specified visits. Assessments planned in this protocol will be according to clinical practice.
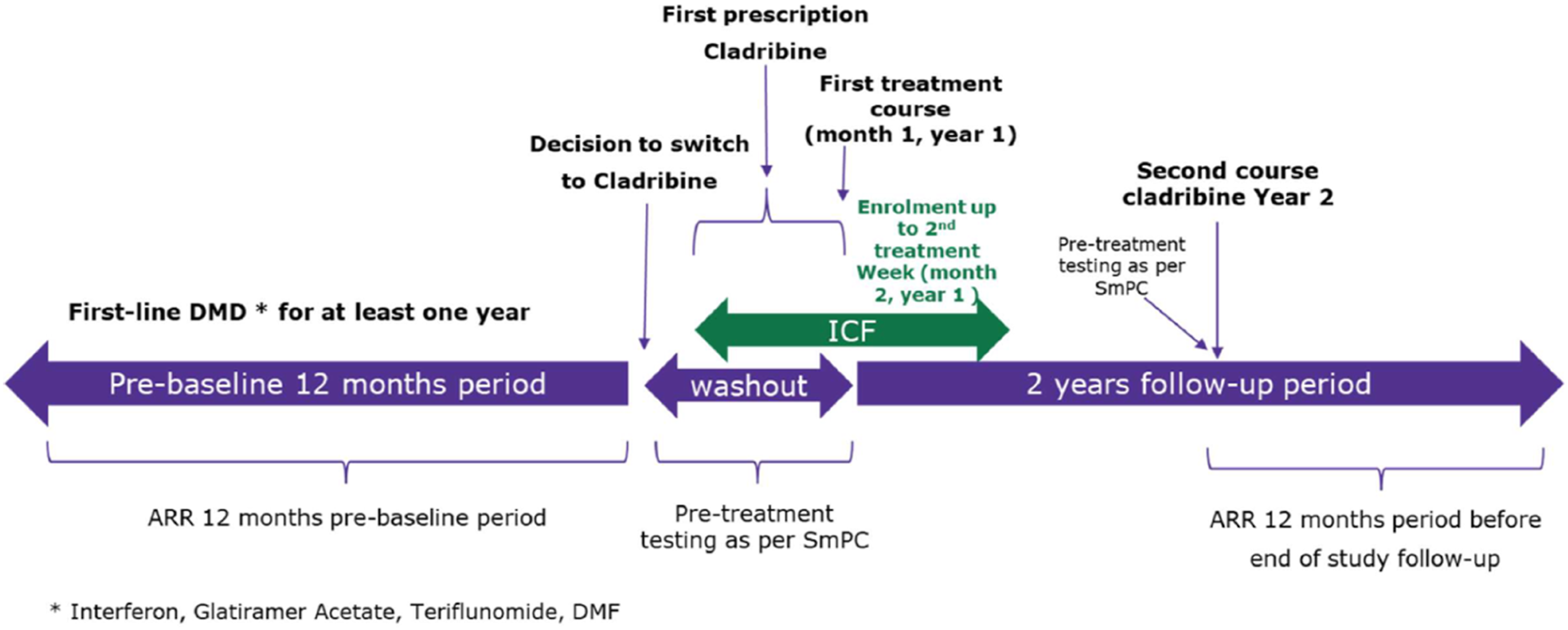
Patients will be enrolled consecutively at study sites in the participating countries. Each patient will be followed for the course of their standard cladribine treatment (approximately 2 years according to clinical practice).
Figure 1 provides a schematic overview of the study design. Figure 2 provides an overview of the patient journey within the study. Enrollment may take place from the time of cladribine prescription and until the second treatment week, in the first treatment year. Schematic of study design. Patients switching to cladribine tablets from first-line DMDs (IFN-b1a, IFN-b1b, glatiramer acetate, dymethylfumarate, and teriflunomide) after at least 1 year of treatment can be recruited over a 30-month period and will be followed up for 24 months. Pts, patients; IFN, interferon; DMF, dymethylfumarate; DMD, disease modifying drug. Study patient journey. Eligible patients receive treatment with cladribine tablets per the SmPC. ARR over the second follow-up year will be compared with the ARR over the 12-month pre-baseline period. ARR, annualized relapse rate.
Schedule of Assessments. 9-HPT, 9-Hole Peg Test; EDSS, Expanded Disability Status Scale; T25FW, Timed 25-Foot Walk.

The first visit will be the baseline visit. Patients who meet all eligibility criteria and who sign the ICF can be enrolled into the study. Inclusion can occur at the time point of treatment prescription with cladribine tablets up until the second treatment week of the first treatment year. Baseline data will be collected at the time of the visit.
In addition to the baseline data, data collection is planned at 4 supplementary visits according to routine practice: at month 6, 12, 18, and 24 approximately.
Outcomes
The primary outcome is the difference in ARR between the pre-baseline 12-month period over the 12-month period before the end of study follow-up (2 years).
Secondary outcomes are the difference in ARR between the pre-baseline 12-month period and over the 12-month period after the start of cladribine (1 year); the percentage of patients with 6-month disability progression at the end of the study follow-up period (2 years), measured by means of the Expanded Disability Status Scale (EDSS), 7 Timed-25 Foot Walk test 8 (T25FW, optional), and 9-Hole Peg test 9 (9-HPT, optional); the percentage of patients with 6-month disability improvement measured at the end of the study follow-up period (2 years) by means of the same tests; occurrence of adverse events and of serious adverse events; quality of life measured via the MS-specific quality of life questionnaire MSIS-29 10 ; treatment satisfaction measured by the TSQM v1.4 11 questionnaire; and data on healthcare resource utilization will be collected by means of the EuroQol EQ-5D-3L 12 questionnaire. Treatment adherence will be assessed as the percentage of cladribine tablets taken vs prescribed dose. Disability progression is defined as progression on ⩾1 of 3 components (EDSS, T25FW, and/or 9HPT) confirmed ⩾24 weeks apart and with a ⩾20% minimum threshold change for T25FW and 9HPT. Progression on EDSS is defined as at least 1 point in the EDSS score or an increase of at least 1.5 points if the baseline EDSS score is 0. Disability improvement is defined as improvement on ⩾1 of 3 components (EDSS, T25FW, and/or 9HPT) confirmed ⩾24 weeks apart and with a ⩾20% minimum threshold change for T25FW and 9HPT. Improvement on EDSS is defined as a decrease in EDSS by at least 1 point (1.5 points if baseline EDSS was 1.5). T25FW and 9HPT are optional and will be performed if done as per routine practice.
The Number of MRI lesions (T2, T1 Gd+, and CUA) at baseline and years 1 and 2 will be evaluated and compared between the pre-baseline 12 months and post-baseline results (years 1 and 2) as exploratory outcomes. An exploratory subgroup analysis will be conducted on effectiveness endpoints, with 5 groups defined according to the pre-baseline DMD treatment (IFN-b1a/IFN-b1b/glatiramer acetate, teriflunomide, and DMF).
Sample Size
Sample size calculation has been based on the study’s primary endpoint, which is the difference in the ARR between the ARR during the 12-month pre-baseline period and the ARR over the 12-month period before end of study follow-up (2 years). In the ONWARD study, 5 where cladribine tablets was added to IFN-beta treatment in patients failing the latter, the additional reduction in the mean ARR exerted by cladribine tablets was 0.20 (from 0.32 in the placebo+IFN-beta to 0.12 in the cladribine tablets 3.5 mg/kg + IFN-beta group). A similar reduction in the mean ARR (from 0.33 in placebo to 0.14 in cladribine tablets 3.5 mg/kg group) was documented in CLARITY trial, 3 which included treatment naïve patients but also patients switching from IFNs or glatiramer acetate (30% of the study population). In a recent post-hoc analysis of CLARITY focusing on high disease activity patients (13), those pre-treated with IFNs or glatiramer acetate afforded a similar reduction in ARR compared to the entire study population, namely, from 0.44 in placebo to 0.25 in cladribine tablets 3.5 mg (difference 0.19). However, ONWARD only included patients pre-treated with interferons or glatiramer acetate and not with teriflunomide or DMF. Therefore, a more conservative approach was taken, by which the study proposed here is powered to detect a clinically meaningful 20% reduction in ARR, also in agreement with published clinical trials on RRMS (14). Thus, on the basis of the effect size reported for the high disease activity, IFN/glatiramer acetate pre-treated population, a sample size of 219 patients is required for a paired t-test to detect a 20% decrease in ARR with 0.05 statistical significance (two-tailed P value) and 80% power using paired t-test comparison. For the calculation of the required sample size, we assumed an average reduction of 20% in ARR, a mean ARR of 0.44 in the pre-baseline period, and a standard deviation of the difference of 0.46. These yielded a Cohen D = 0.19, which was further used in the calculation of sample size. Anticipating a 10% drop-out rate leads to a total sample size of 242 patients, who will be recruited in the participating countries.
Data Management
The data to be collected in the study will be obtained by an electronic Care Report Form (eCRF). The main purpose of the eCRF is to obtain data required by the non-interventional study protocol in a complete, accurate, legible, and timely manner. The data in the eCRF should be consistent with the relevant source documents.
The Investigators or designees are responsible for ensuring that the data collected in the course of this study is accurate and documented appropriately on all applicable forms. Data will then be processed, evaluated, and stored in anonymous form in accordance with applicable data protection regulations. The Investigators must ensure that the eCRFs and any other associated documents forwarded to Sponsor or its designated organization contain no mention of any subject names.
Further to the data that the Investigators will enter to the eCRF in each visit, following history taking, clinical examination, and evaluation of laboratory results, patients will complete the MSIS-29, EQ-5D-3L, and TSQM v1.4 questionnaires at the sites and the information will be transcribed into the eCRF by the Investigator site staff. Pre-baseline data will be obtained from patient records and will be transcribed into the eCRF by the Investigator site staff.
The data will be entered into a validated database. The Sponsor/CRO or its designee will be responsible for data processing, in accordance with the Sponsor’s/CRO’s data management procedures. Database lock will occur once quality control and quality assurance procedures (including SAE/SADR reconciliation) and coding activities have been completed. PDF files of the eCRFs will be provided to the Investigators at the completion of the study.
Data Analysis
Statistical programming and analyses will be performed using SAS 9.4 (or higher). Detailed methodology for summary and statistical analyses of data collected in this study will be documented in a statistical analysis plan (SAP), which will be finalized prior to database lock and will be included in the clinical study report for this protocol. The final SAP will take into account any amendment to the protocol.
All data analyses will be performed on the Full Analysis Set (FAS), defined as all the patients who provided informed consent and who received at least one dose of cladribine. Missing information will be captured for quantitative as well as qualitative variables by the category “Missing” in the summary statistics. If there are no missing values this will be indicated by “0” unless otherwise specified. Due to the longitudinal nature of the data and the lengthy follow-up period, it is possible that missing outcome data will be present. Patterns and degree of missing data will be summarized. No imputation of missing data will be done.
Quality control activities will include verification of data and ensure the data collected in eCRF is accurate, valid, and in accordance with the protocol.
Statistical Methods
All endpoints will be presented by descriptive statistics for baseline, 12 months, and end of study assessments. Assessments done at 6 months and 18 months will be presented similarly. Quantitative (continuous) variables will be summarized with the use of descriptive statistical measures [mean value, standard deviation (SD), median, min-max, and Q1-Q3]. Qualitative (categorical) variables will be displayed as frequencies and percentages (N, %).
The normality of distributions will be examined using the Shapiro–Wilk test in order to determine whether or not to use parametric methods for analysis.
Primary Endpoint: Pre- and post-baseline ARR will be compared by ANOVA or Wilcoxon signed-rank test if data is not normally distributed. The 95% confidence interval (CI) for the difference will be presented.
If appropriate sensitivity analyses will be done to determine the impact of confounding factors or to describe the outcome for special subject groups fulfilling predefined criteria. Details will be described in the SAP. Two specific groups of interest are subjects with a pre-baseline treatment period on a first-line DMD of at least 18 months, vs subjects with smaller treatment periods. The change in pre- and post-baseline ARR will be compared between these 2 groups, to account for the possibility that the full therapeutic effect of the respective first-line DMDs appears with a delay (up to 6 months).
For Secondary and exploratory endpoints, the following calculations and comparisons will be performed: • Pre- and post-baseline ARR (1 year) will be compared by ANOVA or Wilcoxon signed-rank test if data is not normally distributed. The 95% confidence interval (CI) for the difference will be presented. • For all other secondary endpoints, 95% CI will be calculated for the result at each timepoint and for the differences whenever applicable, using appropriate methods. • In the analysis of 6-month disability progression/improvement EDSS, T25FW and 9-HPT values must be obtained at sequential time points, separated by 6-month intervals. As T25FW and 9-HPT are optional and will be performed if done as per routine practice, missing values may occur and could lead to bias in their analysis. Methods to handle this bias issue will be described in detail in SAP. • A subgroup analysis will be conducted on effectiveness endpoints (ARR at 1 and 2 years, percentage of patients with 6-month disability progression and improvement), with 5 groups defined according to the pre-baseline DMD treatment (IFN-b1a/IFN-b1b/glatiramer acetate, teriflunomide, and DMF).
A comparison of these endpoints in subjects with a pre-baseline treatment period on a first-line DMD of at least 18 months, vs subjects with smaller treatment periods will also be performed, similarly to the primary endpoint.
Main analysis will be performed once all the patients completed the study with the purpose of evaluating all endpoints and database is locked. Two descriptive interim analyses will be performed, respectively, when 30% and 60% of the patients have completed the 12-month follow-up visit, to also observe pre-baseline ARR and the variances adherence with sample size assumptions. An alpha adjustment for the multiplicity of the analysis will be introduced if the interim analysis will lead to change in sample size strategy. The interim analyses will be based on a subset of main analysis outputs and the list will be detailed in the SAP.
Monitoring
Risk-based monitoring will be performed for this study. Risk-based monitoring is the process of ensuring the quality of clinical studies by identifying, assessing, monitoring, and mitigating the risks that could affect the quality or safety of a study. It can facilitate efficient trial delivery without compromising patient safety or data quality. A Sponsor or an appointed CRO will identify critical data and processes, performs a risk assessment, and then develops a monitoring plan that focuses on the important and likely risks to critical data and processes.
A Sponsor or an appointed CRO Monitor will mainly perform “remote” monitoring calls at regular intervals.
“On-site” visits may be performed, if required, at any time during the study. For hospitals where potential quality risks are identified, on-site visits can verify that the study is being carried out according to the protocol.
Monitoring on-site visits will involve checking of CRFs against original patient records and identification of any questions or problems related to study conduct or data collection. Investigators must therefore ensure that the Monitor has access to relevant documents during monitoring visits, and that they and/or relevant site staff members are available to discuss any issues that may arise.
The study Monitor will send monitoring reports to the Sponsor.
Auditing
In compliance with regulatory requirements, the Sponsor, a third party on behalf of the Sponsor, regulatory agencies, or Independent Ethics Committees (IECs) may conduct quality assurance audits/inspections at any time during or following a study. The Investigator must agree to allow auditors/inspectors direct access to all study-related documents, including source documents, and must agree to allocate his or her time and the time of his or her study staff to the auditors/inspectors in order to discuss findings and issues.
The protocol, each step of the data capture procedure, and the handling of the data, as well as the study report, may be subject to independent Clinical Quality Assurance. Audits may be conducted at any time during or after the study to ensure the validity and integrity of the study data.
Archiving
The archive should be maintained for the period specified by local regulations, where applicable. All original subject files (medical records) must be stored at the site (hospital, research institute, or practice) for the longest possible time permitted by the applicable regulations. In the absence of applicable regulations, the archive should be maintained for at least 5 years after the final study report or the first publication of study results, whichever comes later. In any case, the Investigator should ensure that no destruction of medical records is performed without the written approval of the Sponsor.
Handling of Adverse Events
The recording period for AEs begins, when the subject is initially included in the study (date of signature of first informed consent) and continues at least end of the mandatory safety follow-up period at last study visit. However, as enrollment may take place from the time of cladribine prescription up until the second treatment week, in the first treatment year, any adverse drug reactions (ADRs) assessed as related to cladribine by the Investigator after the first dose of cladribine but before signature of informed consent must be recorded in the eCRF as well.
All adverse events, as specified above, occurring during the study, must be documented by the Investigator in the eCRF, including its description, seriousness, severity (grading), duration (onset and resolution dates), causal relationship, any other potential causal factors, actions taken with the study drug (e.g., dose reduction, and withdrawal), required treatment, and outcome of the AE.
Death, disability, and hospitalization are considered outcomes in the context of safety reporting and not usually considered ARs/AEs. Therefore, the primary cause of death, disability, or hospitalization should be recorded and reported as an SAE/AR, and the outcome should be recorded in a separate data field. However, a term for the outcome will be selected if it is the only information reported or provides significant clinical information.
If death occurs, the primary cause of death or event leading to death should be recorded and reported as an SAE. “Fatal” will be recorded as the OUTCOME of this respective event and not be as separate event. Only, if no cause of death can be reported (for example, sudden death and unexplained death), the death per se might then be reported as an SAE.
Reports of special situations are also to be recorded in the eCRF following the AE procedure, even if occurring without AE. Special situations, in the context of this study, are the following conditions: malignancy, severe/serious infection, and severe lymphopenia.
Pregnancy or breastfeeding must be recorded in the eCRF and additionally be reported to the Sponsor immediately (within 24 hours of awareness) by using separate paper data collection forms for pregnancy, independent if an AE was reported or not. The outcome of the pregnancy should be followed up and reported to the Sponsor until delivery.
Women who become pregnant under therapy with cladribine tablets should discontinue treatment.
Ethics and Dissemination
Prior to commencement of the study at a given site, the protocol will be submitted together with its associated documents (informed consent form, PROs, etc.) to the responsible IECs for its favorable opinion/approval. The written favorable opinion/approval of the IEC will be filed by the Investigator and a copy will be sent to the Sponsor.
The study must not start at a site before the Sponsor has obtained written confirmation of favorable opinion/approval from the concerned IEC. The IEC will be asked to provide documentation of the date of the meeting at which the favorable opinion/approval was given, and of the members and voting members present at the meeting. Written evidence of favorable opinion/approval that clearly identifies the study, the protocol version, and the subject information and consent form version reviewed should be provided. Where possible, copies of the meeting minutes should be obtained.
Amendments to the protocol will also be submitted to the concerned IEC, before implementation in case of substantial changes.
Key IEC approvals received to date are REC South East Committee (Norway, reference number 31473), Comitato Etico A. O.R.N. (Azienda Ospedaliera di Rilievo Nazionale, Italy, 10/19-10/10/2019), Hospital Garcia de Orta Health Ethics Committee (Portugal, no reference provided), the Bioethics Committee At The Medical University Of Białystok (Poland, R-I-002/391/2019), Comitato etico cantonale (Switzerland, 2020-01892 EC 3714), and Scientific Council of "Ahepa University General Hospital Of Thessaloniki" (Greece, #663).
The International Committee of Medical Journal Editors (ICMJE) authorship guidelines will be followed in the preparation of all publications that will be derived from this study. Professional writers will not be employed.
Informed Consent
An unconditional prerequisite for a subject’s participation in the study is his/her written informed consent. The subject’s written informed consent to participate in the study must be given before any study-related activities are carried out.
Adequate information must therefore be given to the subject by the Investigator before informed consent is obtained (a person designated by the Investigator may give the information, if permitted by local regulations). A subject information sheet in the local language will be provided by the Sponsor for the purpose of obtaining informed consent. In addition to providing this written information to a potential subject, the Investigator or his/her designee will inform the subject verbally of all pertinent aspects of the study (the language used in doing so must be chosen so that the information can be fully and readily understood by laypersons). Depending on national regulations, a person other than the Investigator may inform the subject and sign the informed consent form.
The informed consent form must be signed and personally dated by the subject and the Investigator. The signed and dated declaration of informed consent will remain at the Investigator’s site, and must be safely archived by the Investigator. A copy of the signed and dated information and consent form should be provided to the subject prior to participation.
Whenever important new information becomes available that may be relevant to the subject’s consent, the written subject information sheet and any other written information provided to patients will be revised by the Sponsor and be submitted again to the IEC/IRB for review and favorable opinion. The agreed, revised information will be forwarded to each subject in the study. The Investigator will explain the changes to the previous version.
Patient Identification and Privacy
A unique patient number will be assigned to each subject at inclusion. This number will serve as the subject’s identifier in the study as well in the study database.
The Investigator must ensure that the patients’ anonymity is maintained. On the eCRFs or other documents submitted to the Sponsor, patients should not be identified by their names, but by their assigned identification numbers. If subject names are included on copies of documents submitted to the Sponsor, the names (except for initials) must be obliterated and the assigned subject numbers added to the documents.
The Investigator should keep a separate log of patients’ identification numbers, names, addresses, telephone numbers, and hospital numbers (if applicable). Documents not for submission to the Sponsor, such as signed informed consent forms, should be maintained in strict confidence by the Investigator.
Only authorized persons will have access to identifiable personal details, if required for data verification. The subject’s original medical data that are reviewed at the site during source data verification by the Monitor, audits, and Health Authority inspections will be kept strictly confidential. The Investigator agrees to provide direct access to these documents to the Sponsor and to Health Authority representatives. The Investigator is responsible for retrieving information from personal medical records.
Data protection and privacy regulations will be observed in capturing, forwarding, processing, and storing subject data. Patients will be informed accordingly and will be requested to give their consent on data handling procedures in accordance with national regulations.
Discussion
With a significant number of DMTs for MS available on the market and more becoming available, it is imperative that we understand the patients’ journey through treatments and that we make evidence-based treatment switches possible. 15 Cladribine tablets, approved in late 2017 by the European Medicines Agency 6 and in early 2019 by the US Food and Drug Administration, 16 are among the drugs that were made available to patients and physicians relatively recently. Approximately one-third of patients in the pivotal CLARITY trial switched to cladribine tablets from interferon-beta or glatiramer acetate 3 ; however, neither DMF nor teriflunomide treated patients were enrolled as these treatments were approved after the completion of CLARITY. Real-world cohorts published recently report on patients switching from all first-line treatments, including DMF and teriflunomide, however, in a descriptive manner,17,18 or in very small numbers. 19
CLAD CROSS envisages to collect adequate data, to allow the characterization of the efficacy and safety profile of cladribine tablets when administered in sequence to first-line treatments, both vs the entire group of DMDs as a whole and vs each individual drug. Quality of life, treatment satisfaction, and healthcare resource utilization data that can support pharmacoeconomic analyses will also be collected.
Footnotes
Author contributions
SD conceived of the study.
GT, SD, and NG initiated and implemented the study design.
GT provided statistical expertise in clinical trial design and in conducting the statistical analyses.
All authors contributed to refinement of the study protocol and approved the final manuscript.
Declaration of Conflicting Interests
SD in an employee of Merck AE, Greece. All remaining authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Merck A.E., Greece, an affiliate of Merck.
Protocol Version
2.0, April 7th, 2021
Trial Registration
NCT04934800.