
Abstract
Mutations in dental hypophosphatasia (HPP) have been reported less than those in other types of HPP because the symptoms are mild or the dental lesions are only partial manifestations of other types of HPP. In this case, we observe the clinical manifestation of dental hypoalkaline phosphatase by analyzing the genetic mutation and biochemical parameters in child. The clinical data of the child with odonto HPP were collected and analyzed. The blood samples of the child and his parents were sequenced and verified using Sanger through a specific probe capture and high-throughput second-generation sequencing technology. Major clinical manifestations in the patient were early loss of deciduous teeth, significantly lower serum alkaline phosphatase (ALP) levels, lower active vitamin D, and increased blood phosphorus, but no abnormality was observed in the oral X-ray. Two missense mutations—c.542C>T (p. ser181leu) and c.644 T> C (p.Ile215Thr)—were found in exon 6 of the ALPL gene from the father and mother, respectively. The clinical manifestations of odonto hypophosphatasia were early loss of deciduous teeth and significantly reduced serum ALP levels. Of 2 mutations—c.542C>T (p.ser181leu) and c.644 T> C (p.Ile215Thr)—in the ALPL gene, c.644 T> C (p.Ile215Thr) was a new mutation.
Introduction
Hypophosphatasia (HPP) is a rare genetic disorder, caused by the mutation of the ALPL gene that encodes tissue-nonspecific alkaline phosphatase (TNSALP), resulting in defective bone and tooth mineralization. Very few studies discuss the prevalence of HPP, which is 1 per 300 000 and 16 370 births for severe and moderate HPP in Europe, respectively. 1 Sporadic and monoploid cases are quite rare. 2 The clinical manifestations of HPP vary greatly, ranging from stillbirth without a mineralized bone to mild late adult complications. According to the onset age and severity, HPP can be classified into 6 categories: perinatal lethal, perinatal benign, infantile, childhood, adult, and dental HPP. 3 A child with perinatal HPP is usually diagnosed by the doctor at birth or through an ultrasound before to delivery. Skeletal anomalies, such as malformed chest walls and lengthy bones, are how it presents itself. The X-ray will reveal evidence of hypomineralization, or a drop in mineral concentration, in the bones. With a significant risk of stillbirth or death shortly after birth, this kind of HPP can be lethal. By the time the child reaches 6 months of age, the doctor will diagnose them with infantile HPP. An X-ray can identify its primary features, which include fractures and rickets. The infant’s capacity to develop is hampered by the deficiency of minerals and a malfunctioning metabolic process; this kind of HPP can be lethal. After 6 months of age, children with HPP start to exhibit signs and symptoms. When a youngster doesn’t meet developmental goals for motor skills, the doctor typically makes the diagnosis. Early loss of deciduous teeth, including the root, before the age of 5 years is the most typical sign. This is not the same as typical tooth loss, which occurs from the age of 5 years to the preteen years when the teeth progressively fall out due to root resorption. The adult classification frequently manifests in childhood but is not identified until adulthood. Adult HPP can involve non-specific musculoskeletal conditions, delayed healing, and recurrent femur and metatarsal bone fractures. It shows up as a weakening of the bones, and adults may experience early tooth loss or persistent pain in their joints and muscles. Among them, dental HPP refers to only dental complications in which primary incisors are exfoliated prematurely between the ages of 1 and 4 years without inflammation and other metabolic abnormalities of the skeletal system. 4 To date, mutations in dental HPP have been reported less than those in other types of HPP because the symptoms are mild or the dental lesions are only partial manifestations of other types of HPP.5-8 These patients often present with dental complications without radiological or histopathological evidence of rickets or osteomalacia. As a result, it becomes more crucial to identify the factors which play a major role in detection of HPP. Over 400 pathogenic variants of the ALPL gene have been recorded in the ALPL gene variant database. 9 In this case report, we analyzed the clinical and genomic characteristics of a child with dental HPP along with his family members to improve the understanding of the disease.
Case Description
The proband, a child aged 5 years and 1 month, height 108 cm, weight 17.7 kg, with sitting height 61 cm, was admitted to the pediatric care department of our hospital in March 2021, complaining about 4 years of continual tooth loss. He has a history of deciduous tooth loss (lower incisor, but the details were not clear) after falling accidentally when he was 1 year old; thereafter for nearly 4 years, he was treated in the stomatology clinic of a Grade III hospital for dental repair (use of dental fillings to fix or repair chipped or broken tooth), orthosis (braces), and denture (prosthetic devices to replace missing teeth), but the teeth at the fixed position of the denture were more likely to fall out. During admission, he had lost 10 deciduous teeth and no new permanent teeth have emerged.
Birth history
He was the second child to his mother”, born from natural delivery. His birth weight was 2900 g, and the Apgar score was normal. The childcare department regularly monitored body length, body weight, and the emergence of deciduous teeth at normal levels.
Physical examination
He had clear consciousness, good mental response, mild costal margin eversion, and 10 remaining oral deciduous teeth. His tooth color was normal, with no abnormal oral mucosa. His gingival color was pink, with the eruption of permanent teeth—right lower central incisor I, which was lost during denture and orthosis. He had a soft neck, and the results of his heart, lung, abdominal, and neurological examinations were normal. His limbs could move freely with no deformity, and there was no bracelet or anklet sign (any abnormality’s signs in hands and legs).
Family history
His parents were not inbred with no clinical manifestations such as bone and tooth loss. He had a sister, aged 8 years, who grew normally after birth and had no premature deciduous teeth or bone complications. His grandfather and grandfather’s brother had premature tooth loss, no fracture, fatigue, or bone pain. Moreover, there was no history of childhood growth retardation.
Auxiliary examination
“Bone metabolism tests revealed the low serum alkaline phosphatase (ALP) levels of 37 U/L (reference range: 143-406 U/L) however, blood calcium level was 2.44 mmol/L (reference range: 2.1-2.9 mmol/L) and blood phosphorus level was 1.76 mmol/L (reference range 0.85-1.55 mmol/L). Parathyroid hormone levels were 20.380 pg/mL (reference range: 16.000-65.000 pg/mL) and 25-hydroxyvitamin D3 level was 11.87 µg/L (reference range: 15-51 µg/L) (Table 1). His blood routine test, urine routine test, liver function, kidney function, myocardial enzyme profile, electrolytes, thyroid function, erythrocyte sedimentation rate (ESR), and other trace elements (copper, iron, lead, calcium, zinc, and magnesium) were normal. Furthermore, the karyotype analysis revealed no abnormalities (Figure 1). Additionally, an X-ray of the patient’s upper and lower limbs showed no signs of incomplete bone calcification (Supplemental Figure 1). No obvious abnormalities in the dental film of the oral cavity were observed, and the arrows in the second and third images of the dental film pointed to newly erupted permanent teeth (Figure 1). In addition, the pathology of his deciduous teeth was normal (Figure 2). Low serum ALP levels of 40 and 24 U/L were observed for his father and mother, respectively (reference range of adult male: 45-125 U/L; and reference range of adult female: 35-100 U/L). No abnormality was observed in the oral dental film of his parents.
Biochemical parameters.

Abbreviations: ALP, alkaline phosphatase; Ca, calcium; P, phosphorus.

X-ray of teeth: (a) oral and dental films of the proband at 3 years and 2 months, (b) oral and dental films of the proband at 5 years and 1 month, (c) oral and dental films of the proband at 5 years and 6 month, (d) oral and dental films of the proband’s mother, and (e) oral and dental films of the proband’s Father.

Pathological figures and clinical appearance of the deciduous teeth: (a) deciduous teeth adjunct surface, (b) deciduous teeth buccal lingual surface, (c) deciduous teeth maxillofacial, (d) Uneven enamel thickness, dysplasia, irregular dentin calcification (Mag 10×10), (e) cementum dysplasia, large pulp cavity (Mag 4×10), (f) Uneven enamel thickness, dysplasia (Mag 20×10), (g) local enamel dysplasia of teeth, visible ribbon depression (Mag 20×10).
Genetic testing and results
With the informed consent of his family members, peripheral blood samples were collected from the patients and his family members (father, mother, and sister) with 2 mL of EDTA anticoagulant. Kingland Testing Company was commissioned to conduct chromosome karyotype analysis, whole exon detection, and locus verification, which were completed using the Illumina sequencing platform (Supplemental Figure 2). The average sequencing depth of known gene exons and 5-bp sequences in the upper and lower reaches of the human genome was 90×, and approximately 98% of the target sequences were more than 20×. The secondary analysis was performed using the GATK software suite for sequencing data analysis. Sequencing fragments were compared with the UCSC hg19 reference genome using the Burrows-Wheeler-Alignment Tool (BWA). An analysis of point mutation and small fragment insertion and deletion mutation was also conducted in which variants were annotated using the VEP software (Variant Effect Predictor) and were screened based on ClinVar, OMIM, HGMD, and gnomAD databases.
Proband’s Sanger gene sequencing verification results and peak demonstrated complex heterozygous mutation of the ALPL gene (Figure 3). The progenitor contained 2 gene mutation sites:
(1) Chromosome location chr1:21890603, located at the sixth exon coding region 542 heterozygous mutation C> T (c.542 C> T), resulting in the conversion of amino acid number 181 from serine to leucine (p.Ser181Leu)
(2) Chromosome position chr1:21890705, located at the sixth exon coding region 644 heterozygous mutation T> C (c.644 T> C), resulting in the conversion of the 215th amino acid isoleucine to threonine (p.Ile215Thr)
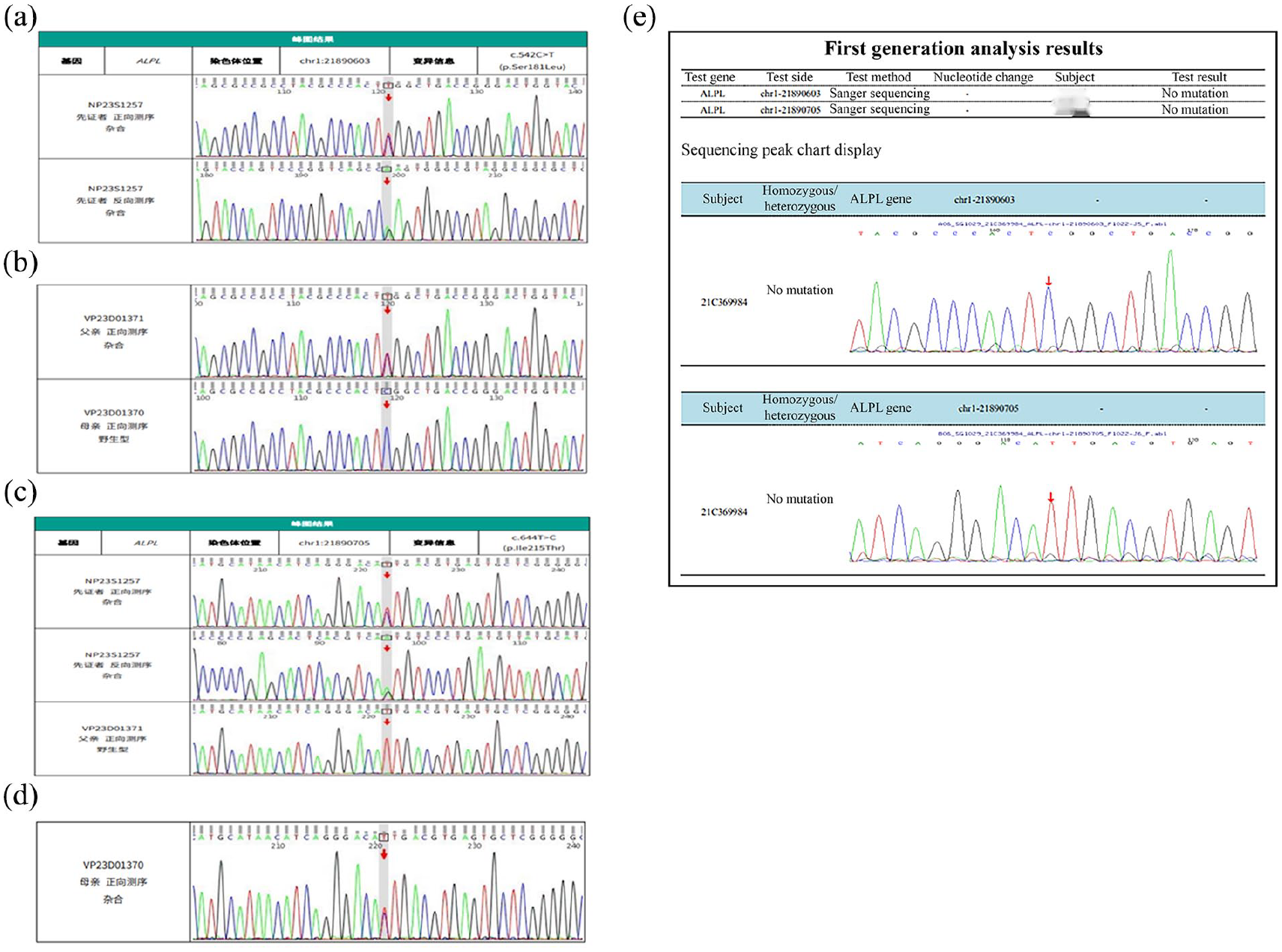
Genetic testing: (a) genetic testing of the proband, (b) genetic testing of proband’s father and mother, (c) genetic testing of the proband and his father and mother, (d) genetic testing of the proband’s mother, and (e) genetic testing of the proband’s sister.
The analysis of the ALPL gene in both parents showed that they were carriers of heterozygous gene mutation. His father had a missense mutation in the gene locus NM_000478.6: c.542 C> T (p.Ser181Leu), which was not recorded in ESP6500siv2_ALL and 1000 Genomes (1000g2015aug_ALL) database but was recorded in dbSNP147 database (rs199590449: Bioinformatics software predicted its therapeutic potential). His mother had a mutation in the gene locus NM_000478.6:c.644 T> C (p.Ile215Thr), which has not been reported yet. No mutation was detected in his sister (Figure 4).

Predicting the structure: (a and b) predicting the structure of proband’s mother’s protein. 215 ILe (green) mutates to Thr, a new hydrogen bond (shown in the red box) formed with the 212 Ile (yellow), which may change the intermolecular force, thus affecting the stable structure and physicochemical properties of the protein. (c and d) predicting the structure of proband’s mother’s protein. 181 Ser (green) mutates to Leu, the 2 hydrogen bonds formed with Asp at 183 (yellow) lost, resulting in weakened intermolecular forces, which may lead to structural instability of the protein and affect its biological function.
Discussion
Dental HPP is an inherited disorder characterized by a decline in serum ALP activity, which leads to inadequate bone mineralization. It is clinically manifested with variable symptoms including rickets such as bone changes, low bone mineral density, short stature, muscle weakness, craniosynostosis, and premature loss of deciduous teeth. 10 In this case, the patient began to have deciduous tooth loss at the age of 1 year and was treated in the oral clinic with braces and other fixed teeth; however, it was not effective. His serum ALP and 25-hydroxyvitamin D3 levels were 37 U/L significantly decreased, blood phosphorus level was increased, and blood calcium level, along with parathyroid hormone, was in the normal range. In addition, changes in bone metabolism indexes were consistent with abnormal HPP bone mineralization. There was no calcification incomplete phenomenon in the upper and lower limbs. It is known that the number of premature deciduous teeth lost by children paralleled the severity of their overall HPP disease. 11 Amorphous pyrophosphate is an irreplaceable regulator of the development of acellular cementum in the tooth roots and a key factor in determining the hard-soft interface between the periodontal membrane and cementum. Due to the low activity of ALP and the high levels of abiotic pyrophosphate, acellular cementum dysplasia or even loss may affect the development of tooth root and periodontal membrane attachment, which eventually leads to the premature loss of deciduous teeth. 12 Therefore, the lost teeth are usually painless and bleeding-free with roots intact. In this case, the proband had multiple deciduous teeth with intact roots (Figure 2). As mechanized pyrophosphate plays an important role in the development of acellular cementum, dental tissue is more sensitive to changes in serum ALP levels than bone tissue, 12 which may explain the most common manifestations of HPP. In addition, enamel damage, enlarged root canals and pulp compartments, and reduced and enlarged dentin tubules can also be observed in affected teeth. In this case, no expansion of the root canal and pulp chamber was found in the dental film. The lost intact deciduous tooth was sent to pathology, and the results were normal. Premature loss of deciduous teeth can also lead to alveolar bone defects, dislocation, or premature loss of permanent teeth 13 and even can affect language and nutrition. This shows the importance of professional dental care.
HPP is also associated with mutations of the ALPL gene that is located at 1P3.12, contains 12 exons, and is about 50 kb in size. 14 Mutations related to HPP are found at all exons and mostly comprise missense mutations; however, frameshift and intron mutations were also associated with HPP due to the insertion or deletion of bases. 15 In this case, genetic testing was performed on the proband and his family members to further reveal the etiology and genetic analysis.
The results of gene sequencing showed that the proband carried 2 mutated genes, 1 from the father and 1 from the mother, both of which were missense mutations on exon 6. Father had a mutation in gene locus NM_000478.6:c.542 C> T(p.Ser181Leu), which is expected to change the 181st amino acid of the encoded protein from serine to leucine. Bioinformatics software (rs199590449) and dbSNP147 database predicted the possibility of disease. The same mutation has been reported in 2 cases in patients with HPP 16 : one was a 4-year-old girl with perinatal nonfatal disease and the other was a 15-year-old girl with typical clinical and radiological features of childhood HPP. In addition, low serum ALP activity of the patient and parents was confirmed using molecular analysis of the ALPL gene.17,18 Proband’s mother had a mutation in gene locus NM-000478.6:c.644 T> C (p.Ile215Thr), which is expected to change the 215th amino acid of the encoded protein from isoleucine to threonine. This mutation was not recorded in ESP6500 siv2_ALL, 1000g 2015 aug_ALL, and dbSNP147 databases; however, bioinformatics software predicted that it has a high pathogenic potential. It has been reported that other variant forms in the same location were detected in patients with HPP c.643A>G (p.Ile215Val). 19 The gene test analysis of Proband’s sister showed no related mutation.
Both parents of the proband had decreased serum ALP levels with no skeletal complications. His sister is currently asymptomatic, and genetic testing had not revealed any genetic mutations and her ALP levels were normal. The ALPL gene of Proband’s mother is a new mutated gene. The mutation of 2 genes c.542 C> T(p.Ser181Leu) and c.644 T> C (p.Ile215Thr) inferred that it was associated with premature deciduous tooth withdrawal in the proband Proband’s braces were removed after diagnosis, and no tooth loss occurred again. At present, no abnormal growth and development were found in the patient during the follow-up.
Conclusion
According to this case, c.644 T> C is a new mutation related to HPP, which expands the ALPL gene mutation spectrum. This case will help to enhance the understanding of HPP and positively impact children’s quality of life suffering from HPP.
Supplemental Material
sj-pdf-1-pdi-10.1177_11795565241256615 – Supplemental material for Hypoalkaline Phosphatemia Dental Type: A Case Report
Supplemental material, sj-pdf-1-pdi-10.1177_11795565241256615 for Hypoalkaline Phosphatemia Dental Type: A Case Report by Weihua Liu, Xiaoyang Min, Hongli Wang, Qianqian Lu, Lulu Li and Haiping Chu in Clinical Medicine Insights: Pediatrics
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.