
Abstract
Primary immune deficiency (PID) is a large group of diseases characterized by defective immune function, leading to recurrent infections, and immune dysregulation. Clinical presentations, severity, and complications differ for each disease, based on the components of the immune system that are impacted. When patients with PID present with respiratory symptoms, infections should be initially suspected, investigated, and promptly managed. However, non-infectious complications of PID also frequently occur and can lead to significant morbidity and mortality. They can involve both the upper and lower respiratory systems, resulting in various presentations that mimic infectious diseases. Thus, clinicians should be able to detect these conditions and make an appropriate referral to an immunologist and a pulmonologist for further management. In this article, we use case-based scenarios to review the differential diagnosis, investigation, and multidisciplinary treatment of non-infectious pulmonary complications in patients with primary immune deficiencies.
Keywords
Introduction
Primary immune deficiencies (PIDs) are rare disorders characterized by loss or dysregulation of the immune system leading to a pattern of recurrent infections, which can be severe, life-threatening, and/or difficult to treat. There are more than 400 types of PIDs that vary in severity and the extent of the immune system’s involvement. 1 Pulmonary complications pose a substantial risk of morbidity and mortality in individuals with PIDs. These complications can impact either the upper airways, leading to conditions like otitis media and sinusitis, or the lower airways and lung tissue, resulting in pneumonia, interstitial lung diseases, and bronchiectasis. These complications can be chronic and debilitating. 2 Multidisciplinary care and close monitoring are required by primary care providers, clinical immunologists, and pulmonologists. In this review, we will use a case-based discussion, highlighting the clinical presentation, diagnosis, and management of pulmonary complications in patients with PIDs. We will focus on non-infectious pulmonary complications, including obstructive and restrictive lung diseases, post-infectious complications such as bronchiectasis, and interstitial lung diseases.
Antibody Deficiency Disorders
Antibody deficiency is the most common form of primary immunodeficiency, found in over 50% of individuals with the conditions.
1
These humoral defects can be related to absent B cells, classically seen in X-linked agammaglobulinemia, or reduced antibody production and inadequate antibody responses to antigens, as seen in common variable immune deficiency (CVID). Generally, patients with antibody deficiency are at increased risk of recurrent sinopulmonary infections, which can result in pulmonary complications such as interstitial lung diseases and bronchiectasis
2
A 15-year-old patient with common variable immune deficiency (CVID) presented for a routine physical examination. The patient was diagnosed with CVID at 10 years of age and started on intravenous immunoglobulin (IVIG) infusions. He had a history of wheezing and had been on bronchodilators and inhaled corticosteroids prior to the diagnosis of CVID. He also had pneumonia twice at 10 and 13 years of age. The patient reported no symptoms. His physical examination was within normal limits. The chest examination was clear to auscultation and there was no digital clubbing. However, his pulmonary function test (PFT) revealed a slight decrease in forced vital capacity (FVC) and forced expiratory volume in 1 second (FEV-1) with a normal FEV-1/FVC ratio compared to the previous year. This patient’s computerized tomography (CT) showed ground glass opacities, lung nodules, and basilar septal thickening, as seen in Figure 1. There was no classic appearance of a signet ring sign, peripheral tapering of airways, airway wall thickening, or mucus plugging that would be consistent with bronchiectasis. Splenomegaly was noted on the CT scan which was not appreciated by physical examination. The patient’s complete blood count (CBC) with differentiation was within normal limits. As our patient did not have respiratory symptoms, early interstitial lung disease (ILD) or Granulomatous/lymphocytic interstitial lung disease (GLILD) was possible. Infectious etiologies were ruled out by flexible bronchoscopy and bronchoalveolar lavage. Flexible bronchoscopy and bronchoalveolar lavage (BAL) showed normal lower airway anatomy, with discrete mucoid droplets throughout lower airways which were easily suctioned. Cultures were negative for bacterial, mycobacterial, viral, and fungal pathogens. Thus, non-infectious pulmonary complications are of concern. PFT reported an FVC of 78% predicted, FEV1 of 85% predicted, and FEV1/FVC of 95% predicted, showing a mild restrictive pattern. The diffused capacity for carbon monoxide (DLCO) also slightly declined. Thus, the differential diagnosis at this point included ILD, GLILD, lymphoproliferative lesions, and lymphoma. Other types of interstitial lung diseases including sarcoid-like granuloma, organizing pneumonia, lymphocytic interstitial pneumonitis, and nonspecific interstitial pneumonia should also be considered. Lymphoma was less likely without constitutional symptoms and a normal CBC. A lung biopsy was performed and reported a heterogenous pathology of interstitial pneumonitis, minute granulomas, and lymphocytic bronchiolitis. Based on these findings, he was diagnosed with GLILD. The patient was started on daily prednisone by mouth, followed by mycophenolate and eventually rituximab infusions. A repeat high-resolution chest tomography (HRCT) was performed 8 months after initiating the treatments which showed significant interval improvement of the lesions suggestive of good response to immunosuppressive therapy. There were persistent mild residual nodular and ground-glass opacities predominantly in the lung bases, with interval improvement in the nodular densities in the bilateral lung bases, and with interval decrease in size of the mediastinal, paratracheal, and subcarinal lymph nodes.

HRCT of the patient with GLILD prior to starting treatment, showing a lung nodule (black arrow) and septal thickening (black arrowhead).
CVID is a group of diseases characterized by hypogammaglobulinemia, low levels of immunoglobulin (Ig) A and/or IgM, impaired production of specific antibodies, and recurrent infections. It typically affects patients in late adolescence or early adulthood. 3 Most patients present with an increased frequency of infections, particularly respiratory infections. The estimated prevalence of CVID is approximately 1 in every 25,000 people. 4 While the majority of CVID cases are sporadic, around 20% to 30% of the patients have specific monogenic defects associated with CVID. CVID patients with these monogenic defects tend to have early-onset disease and a family history of CVID. 5 Patients with monogenic causes of CVID frequently experience lymphoproliferative diseases, including GLILD, autoimmune disease, and B-cell lymphopenia. With the ubiquitous use of immunoglobulin replacement therapy (IGRT) in CVID, infectious complications related to antibody deficiencies have been minimized. However, non-infectious complications, specifically chronic lung diseases, develop despite appropriate IGRT and impact prognosis in these patients.6,7 The management of CVID involves immunoglobulin replacement, which can be given either intravenously (IVIG) or subcutaneously (SCIG). The usual initial dose for IVIG is 500 mg/kg every 4 weeks. Generally, IgG trough levels are aimed to be ⩾800 to prevent pulmonary complications in patients with antibody deficiency. 8
Clinical presentation and surveillance of pulmonary complications in patients with antibody deficiency
Patients affected by antibody deficiencies commonly have a history of recurrent pneumonia. These infections may be severe and complicated, requiring intravenous antibiotics and hospital admissions. Some patients may have a history of prolonged hospitalizations requiring supplemental oxygen. Pulmonary complications in patients with antibody deficiency may present as chronic cough, exertional dyspnea, and occasionally, hemoptysis. Physical examination may reveal signs of poor growth, weight loss, digital clubbing, and crackles on lung auscultation. Hypoxemia is usually a late sign in patients with pulmonary complications. Apart from pulmonary infections, which are frequently observed and have to be treated promptly, non-infectious pulmonary conditions should also be considered in the differential diagnosis.
Many patients with pulmonary complications related to their PIDs, especially those in the initial phase of the disease, can be asymptomatic, as observed in case 1. These patients’ physical examinations can be normal. Therefore, routine screening for pulmonary complications is recommended and necessary. Patients with CVID or agammaglobulinemia should have at least 1 baseline HRCT and undergo a pulmonary function test, including lung volumes and DLCO, at least annually.8,9 In selected cases with suspicion of non-infectious pulmonary complications, exercise testing, to assess for hypoxemia or a decrease in lung function with exercise, and blood gases to assess gas exchange may be needed. 10 Chest X-ray is not very sensitive in screening for pulmonary complications. HRCT is very sensitive and is the preferred modality for diagnosing pulmonary complications in patients with antibody deficiencies. HRCT should be obtained when there is clinical suspicion of lung complications and for the purpose of monitoring disease progression. 11 However, patients with CVID are at increased risk of developing lymphoma and caution should be exercised to minimize radiation exposure.
Differential diagnosis of pulmonary complications in patients with antibody deficiency
Infection
For those with antibody deficiencies who present with respiratory symptoms or abnormal radiologic findings, it is important to rule out pulmonary infection first. These patients may not have a fever or appear ill. Inflammatory markers may not be elevated, even during active infection. Serology tests are not practical in those with antibody deficiencies as they are unable to produce antibodies. Pathogen identification relies on polymerase chain reaction (PCR) tests, antigen detection, and cultures. Detailed discussions on types of infection, susceptibilities, and treatment are beyond the scope of this review.
Bronchiectasis
Bronchiectasis is defined as a pathologic condition of the conducting airways characterized by chronic productive cough and radiographic evidence of bronchial dilation. 12 It is the consequence of recurrent infectious or other airway insults, such as aspiration. Patients with antibody deficiency, with a tendency for recurrent pneumonia, are at increased risk for bronchiectasis. This is due to repeated, persistent, or severe infection leading to airway inflammation, regeneration, and eventually structural damage. 13 Recurrent hospitalizations with severe pneumonia (e.g., longer hospital stay or need for supplemental oxygen) elevates the risk of developing bronchiectasis in the later stages of the disease. 14 A cohort study found that the incidence of bronchiectasis was significantly higher in CVID patients with B-cell lymphopenia, compared with those without. 15 The typical history would include a chronic wet-sounding cough, shortness of breath during physical exertion, frequent lower respiratory tract infections, and occasionally hemoptysis. Physical examination may be normal, though they may present with poor growth, digital clubbing, and crackles. Hypoxemia is a late sign and is usually seen in advanced cases.
In patients with bronchiectasis, spirometry may be normal in milder diseases but usually shows an obstructive defect. In some patients, a combined restrictive and obstructive defect may be seen, possibly due to progressive bronchiectatic changes. 16 The best and most sensitive imaging modality for the diagnosis of bronchiectasis is HRCT. Chest radiographs are not very sensitive for diagnosis. The classic finding on HRCT with bronchiectasis is the signet ring sign in which the internal diameter of the bronchus appears larger than that of the adjacent vessel. Other classic findings on HRCT include failure of the airways to taper peripherally, airway wall thickening, and mucus plugging. 17
Interstitial lung disease (ILD)
ILD occurs in approximately 10% to 20% of individuals diagnosed with CVID. 18 In nearly all cases, it is accompanied by lymphadenopathy and splenomegaly.19 -21 These patients can have varied clinical presentations, from being asymptomatic to having significant respiratory insufficiency. When present, symptoms may include a chronic cough and dyspnea with activity. They may have fine crackles on physical examination, although the examination may be normal in the early stages.
In patients with ILD, PFTs may be normal in the initial stage and may show a restrictive defect with or without an obstructive component. Reduced DLCO and impaired oxygenation during exercise can be early markers of ILD. HRCT is the preferred method for diagnosing ILD, as chest radiographs often yield inconclusive results. HRCT features vary from pulmonary nodules, ground-glass opacities, diffused consolidation, granuloma, and fibrosis. 22 PFTs should be regularly obtained and monitored in the follow-up of CVID patients with ILD. 18
ILD has a broad spectrum of disease and has been categorized by various histopathological findings from lung biopsies. 23 The ILD in patients with immunodeficiency most commonly manifested as benign lymphoproliferative pathology with follicular bronchiolitis, nodular lymphoid hyperplasia, and lymphocytic interstitial pneumonia (LIP). Organizing pneumonia and granulomatous inflammation are also frequently seen. 11
When GLILD is suspected, current diagnostic recommendations include pulmonary function testing, chest CT, bronchoscopy, and a lung biopsy. 24 HRCT findings of GLILD pattern include poorly defined areas of nodular consolidation and ground-glass opacity. These patterns often appear predominantly in the lower zones and exhibit an axially bronchovascular or diffuse distribution. Additionally, interlobular septal thickening, and mediastinal, hilar, and/or axillary lymphadenopathy are also observed.18,28 Magnetic resonance imaging (MRI) can be a viable alternative modality, as it does not pose the risk of radiation exposure found in HRCT. However, its lower resolution can potentially reduce the sensitivity of identifying small nodules. Bronchoscopy and microscopic examination and culture of BAL should be done to exclude infection. Additionally, in GLILD, BAL differential cell count will yield an increase in lymphocytes. 29 Pulmonary function tests can show a restrictive pattern. 28 Lung biopsy in GLILD yields heterogeneous pathology of both granulomatous and lymphoproliferative histopathologic patterns. Due to GLILD’s multisystemic presentation with non-caseating granulomas on histology, it can be confused with sarcoidosis, which is relatively rare in pediatric population but can be considered a differential diagnosis in adolescent patients. However, sarcoidosis can be distinguished by normal or high serum immunoglobulin levels, spontaneous remission, and upper lung zone predominance of the disease, as listed in Table 1.11,18,30,31
Distinguishing features between CVID with GLILD and sarcoidosis.
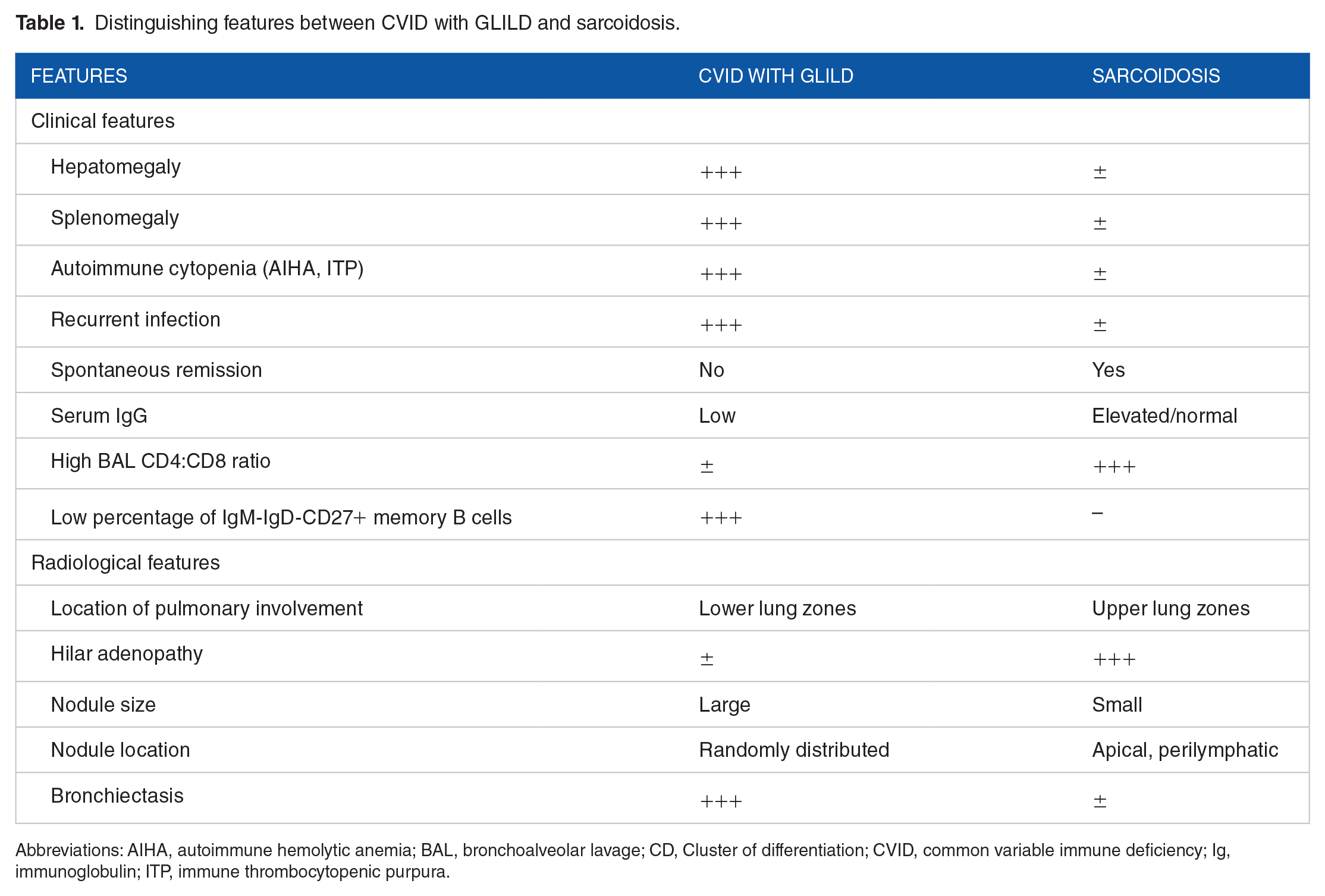
Abbreviations: AIHA, autoimmune hemolytic anemia; BAL, bronchoalveolar lavage; CD, Cluster of differentiation; CVID, common variable immune deficiency; Ig, immunoglobulin; ITP, immune thrombocytopenic purpura.
Other forms of ILD noted in patients with CVID include organizing pneumonia (OP), non-specific interstitial pneumonitis (NSIP), and LIP. However, these histological features commonly coexist, even in the same specimen. 18 Distinguishing features between bronchiectasis, ILD, and GLILD are shown in Table 2.
- Organizing pneumonia (OP) refers to a lung-tissue repair pattern that occurs after injury. It can either be idiopathic or a response to a specific lung injury. Cryptogenic organizing pneumonia (COP) has no identifiable cause and is categorized as idiopathic interstitial pneumonia. It was previously called bronchiolitis obliterans organizing pneumonia and it is often misdiagnosed. However, with appropriate treatment, it has a high rate of recovery. 32 Secondary forms of organizing pneumonia are associated with identifiable causes such as viruses, drugs, inhalation injury, radiation exposure, or malignancy. It is also seen in patients with connective tissue disorders, aspiration, or as a complication of transplantation. 33 Clinical manifestation of OP, similar to other forms of ILD, includes subacute cough, dyspnea, tachypnea, and fine crackles on physical examination. The DLCO is usually reduced and the imaging commonly shows bilateral alveolar opacities and patchy ground-glass consolidation. 34 Lung biopsy reveals intra-alveolar plugs of granulation tissue with connective tissues and myofibroblasts. 23
- Non-specific interstitial pneumonia (NSIP) is characterized by a homogenous interstitial enlargement from the extracellular matrix, inflammatory cells, and fibroblast accumulation. 18 NSIP is characterized by a homogenous interstitial enlargement from the extracellular matrix, inflammatory cells, and fibroblast accumulation. 17 Clinical manifestations are non-specific. It can occur either as an idiopathic condition or as a secondary response to connective tissue disease, exposure to toxins, or other underlying causes. Idiopathic NSIP is a rare diagnosis and requires the exclusion of other potential causes. Patients typically present in their adulthood with shortness of breath, cough, and constitutional symptoms such as fatigue and fever. The condition is more prevalent in females and more than 50% of patients have never smoked. Physical examination may show digital clubbing, crackles on auscultation, and mild resting hypoxemia. Pulmonary function tests may show restriction and a low diffusing capacity for carbon monoxide. HRCT can reveal subpleural reticular changes, traction bronchiectasis, and ground-glass opacities, predominantly in the lower lobe. Honeycombing, which significantly overlaps with other forms of ILD, is occasionally observed. 35
- Lymphocytic interstitial pneumonitis (LIP) is a disease progression from the pulmonary lymphoid hyperplasia around the bronchi, so-called follicular bronchiolitis, to the involvement of the interstitial space, resulting in the alveolar septum expansion. 11 Apart from primary immunodeficiency, LIP is associated with autoimmune diseases, HIV, and hypo/hypergammaglobulinemic states. 36 Patients may present with cough, dyspnea, and chest pain, and physical examination can reveal both signs of consolidation such as bibasilar rales and obstructive patterns with decreased breath sounds and wheezing. 37
Clinical characteristics and investigation findings of bronchiectasis, ILD, and GLILD.

Abbreviation: HRCT, high-resolution chest tomography.
Lymphoma
Patients with CVID are at 30 times increased risk for lymphoma, most commonly non-Hodgkin’s lymphoma (NHL), compared to the normal population. 38 Pulmonary mucosa-associated lymphoid tissue (MALT) lymphomas can be seen in CVID patients.39,40 This can present as pulmonary nodules or masses. Patients may have no respiratory symptoms or may have a cough, tachypnea, difficulty in breathing, wheezing, stridor, or hemoptysis. 41 Lymphadenopathy, hepatosplenomegaly, cytopenia, and constitutional symptoms, such as fever, weight loss, and night sweats are suggestive of lymphoma and should prompt further evaluation by hematology/oncology specialists.
Management of Pulmonary Complications in Patients With Antibody Deficiency
Management of pulmonary complications in antibody deficiency focuses on airway clearance and interrupting the cycle of inflammation and infection. Airway clearance is achieved by using an inhaled solution such as 7% hypertonic saline with chest physiotherapy (CPT). 42 Examples of CPT methods include using an oscillatory positive expiratory pressure (PEP) device, high-frequency chest wall oscillation (HFCWO) autogenic drainage, active cycle breathing with huff coughs, and manual chest percussion. 43
In antibody-deficient patients with bronchiectasis, a double-blind, randomized controlled trial reported that low-dose azithromycin prophylaxis could reduce exacerbation episodes, antibiotics use, and hospitalization. 44 However, it is important to consider the potential risk of bacterial resistance associated with prolonged use of macrolide. 45 In patients colonized with Pseudomonas aeruginosa, inhaled antibiotics such as inhaled tobramycin have been shown to reduce the density of the organism in sputum, as well as the risk of acute exacerbation. 46
In patients diagnosed with GLILD, optimizing trough IgG with the standard dose of IGRT is advised since there is no evidence that increasing the dosage can directly impact GLILD progression. 24 In CVID patients, maintaining serum IgG above 500 mg/dL is reported to prevent infections, while a level above 800 mg/dL can improve pulmonary outcomes. Previously, experts suggested a target trough IgG level of 300 mg/dL above the patient’s pretreatment level. 47 However, the more recent review stated that there are more data supporting individualized dosing based on clinical outcome, rather than merely targeting a trough level. 48 Regardless, despite the optimal immunoglobulin therapy, it is reported that the pulmonary disease may still progress, and additional treatments with immunosuppressive therapy may be needed. 49 In patients who developed worsening respiratory symptoms, decline in PFT (⩾10% decrease in FVC or DLCO), or worsening of HRCT, immunosuppressants are generally required. Oral steroids are usually initiated first, 24 followed by rituximab alone 50 or with azathioprine or mycophenolate in non-responsive cases.51 -54 Some patients have been reported to benefit from tumor necrosis factor (TNF) antagonists, cyclosporine, sirolimus, and abatacept.55,56
This case highlights the value of monitoring pulmonary complications in patients with antibody deficiency, namely CVID, and agammaglobulinemia, on a regular basis. Patients may be asymptomatic early on with underlying lung changes that can be revealed with HRCT. The cornerstone of the treatment of CVID is immunoglobulin replacement to prevent infections, hence preventing pathologic changes such as bronchiectasis, as well as improving pulmonary outcomes. The trough IgG levels of ⩾800 mg/dL in combination with individualized levels based on infections through the course of the disease should be maintained. Additional immunosuppressive therapy, with corticosteroid as a first-line treatment, should be considered in patients who develop pulmonary complications, especially in those who have symptoms or deteriorating lung function. Additionally, these patients should maintain good pulmonary hygiene with chest physiotherapy to prevent further damage to the lungs.
Phagocyte Disorder
Phagocytes are a part of the first line defense of the immune system by ingestion and destruction of pathogens. These disorders involve defects in cell movement, recruitment, activation, or intracellular killing.
A 15-year-old patient with chronic granulomatous disease (CGD) presented with a history of fever and cough. He was seen by his primary care physician initially and treated with oral amoxicillin and azithromycin. However, after 5 days of treatment, his condition did not resolve. His chest x-ray revealed left lower lobe consolidation, which was consistent with pneumonia. He was then hospitalized and treated with intravenous ceftriaxone and clindamycin. Despite a week of intravenous antibiotics, his symptoms persisted. The sputum culture sent before the initiation of intravenous antibiotics came back negative. A chest CT was ordered to evaluate persistent pneumonia. It revealed extensive zones of consolidation with scattered associated adjacent nodules in the anterior and posterior lung bases (Figure 2). Bronchoscopy showed thickened erythematous mucosa. Bronchoalveolar lavage was done to rule out infection. While awaiting culture results, intravenous antifungal therapy was added. However, the patient did not respond. When all cultures came back negative, pulmonary granuloma was suspected. Malignancy is less likely due to the absence of constitutional symptoms and the lesion appearance on the CT. A lung biopsy was done to confirm the diagnosis. A short pulse of prednisolone was given while awaiting the biopsy result, and optimal antibiotic and antifungal treatment were continued throughout the course. The patient’s respiratory symptoms improved, and the consolidation and atelectasis also improved on the follow-up CT scan. The histopathology revealed non-caseating granuloma with scattered microabscesses without organism identified.

HRCT of the patient with scattered granuloma at lung bases (black arrow).
CGD is caused by genetic mutations in the genes encoding the phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidation complex, which plays a crucial role in the production of reactive oxygen species in order to kill ingested microbes. The diagnosis is made by testing for superoxide production by dihydrorhodamine (DHR)-123 flow cytometry assay, and historically by nitroblue tetrazolium reduction. Patients typically present with recurrent infection of catalase-positive organisms such as Staphylococcus aureus, Burkholderia cepecia complex, Serratia marcescens, Nocardia spp, and Aspergillus spp. Commonly affected organs included lungs, lymph nodes, skin and soft tissues, bones, and liver. 57 Apart from infectious complications, CGD is also associated with various autoinflammatory conditions affecting the gastrointestinal tract, lungs, urinary tracts, eyes, and skin. Autoimmune/rheumatologic diseases such as discoid lupus, rheumatoid arthritis, dermatomyositis, sacroiliitis, idiopathic thrombocytopenia, autoimmune hepatitis, and Raynaud phenomenon are also reported in CGD patients.58,59
Respiratory complications, both infectious and non-infectious, are frequently found in CGD patients, affecting 40-85% of patients. Therefore, annual pulmonary examination, pulmonary function test, and chest x-ray are suggested in adult CGD patients. A thoracic CT is recommended every 2 years or when a respiratory event occurs. 60 The findings are often non-specific, ranging from random nodules, ground-glass opacities, focal consolidations, masses, to cavities and abscesses. 61
Differential diagnosis of pulmonary complications in patients with phagocyte disorders
Infection
Some bacteria are reported to cause pneumonia in CGD patients, including Staphylococcus species, Burkholderia cepacia, Nocardia species, Serratia species, and Mycobacterium.58,62 After appropriate cultures and infectious workup are completed, empiric antibiotics should be tailored to cover likely causative organisms. If the patient fails to respond, empiric antifungal should also be considered, as Aspergillus spp most commonly causes pneumonia in CGD.58,62 In this group of patients, invasive Aspergillus infection can occur without any preexisting lung damage, resulting in absence of cavity lesions in most cases. 63
Mulch pneumonitis is a condition caused by extensive inflammatory responses to fungal elements. Patients present with fever, cough, dyspnea, and hypoxia after massive inhalation of fungal spores and hyphae, usually upon exposure to mulch, hay, leaves, and wood chips. 64 Chest X-ray can reveal rapidly progressing bilateral infiltrates, which can overlap with other hypersensitivity pneumonitis syndromes. 65 The diagnosis is made by positive culture or fungal elements in BAL or lung biopsy specimens. Causative organisms are typically Aspergillus species, but Rhizopus and Penicillium species are also concurrently identified.64,66,67 Early treatment with antifungal agents and high-dose corticosteroid to suppress the inflammation must be given since it is associated with high mortality rates. 64
Granulomas
Granuloma formation is a common inflammatory manifestation in CGD, affecting multiple organs including the bowel, brain, spleen, liver, and lungs. The pathogenesis is still unclear. Granulomas are mainly formed by multinucleated giant cells and are typically sterile, although the presence of infection can be difficult to exclude. Lung granulomas can present as airway obstruction and atelectasis, depending on the location of the lesion. Radiologic pulmonary findings can be non-specific, showing consolidation, ground-glass opacities, or pulmonary nodules. 65 Biopsies are often needed to confirm the diagnosis, as well as to rule out an infectious etiology. Histopathologic features are non-specific to CGD, typically showing suppurative, necrotizing, or non-necrotizing granuloma with occasional foci of necrosis, fibrosis, and abscesses. 68
Malignancy
Weel et al 69 reported an increased malignancy risk in CGD patients in 1996 and proposed a hypothesis of cytotoxicity deficit against cancer cells, but no direct association between NADPH oxidase activity and cancer has been found to date. However, cases of Hodgkin lymphoma, acute lymphoblastic leukemia, cervical cancer, colon cancer, pancreatic cancer, and testicular teratoma in CGD patients are reported.70 -72
Non-infectious pulmonary manifestations that are less frequently found in CGD patients include interstitial lung disease, pleural effusions, and chronic obstructive pulmonary disease. 65
Management of pulmonary complications in patients with phagocyte disorders
When a pulmonary lesion is suspected, thoracic CT is the investigation of choice to initially assess the disease. Bronchoscopy is also advised to collect specimens for microbiological analysis. Empiric antibiotics should be given while awaiting the culture results. If no pathogen is identified, non-infectious conditions should be considered, and a lung biopsy can be helpful to determine the diagnosis. Immunomodulator therapies should be discussed, along with empiric antibiotics and antifungal treatment. 60
Established clinical guidelines for managing inflammatory lung disease in CGD patients are currently limited. Corticosteroids are widely used as a treatment of choice, with some reports on thalidomide, hydroxychloroquine, methotrexate, intravenous immunoglobulin, mycophenolate mofetil, cyclophosphamide, and rituximab.60,73 Some experts suggested short courses of high-dose corticosteroid as an initial treatment, and escalating to additional immunomodulators if the patient poorly responds to steroids. Additionally, after the therapeutic antimicrobial course finishes, optimal antibacterial and antifungal prophylaxis should be continued throughout the course of immunomodulators. 65 Infliximab, a TNF-alpha inhibitor, is suggested in CGD-related colitis patients who are steroid refractory. 74 However, associations with serious bacterial and fungal infections were reported so it should be used with caution. 75
Conclusion
Pulmonary complications are common in patients with primary immunodeficiency. Apart from frequent infection, non-infectious pulmonary events are commonly reported as a significant cause of morbidity and mortality. Patients with CVID or agammaglobulinemia should have a baseline HRCT and obtain annual PFT with lung volumes and DLCO, to screen for respiratory complications. If there are new respiratory symptoms or abnormal screening results, bronchiectasis, ILD, and malignancies should be in the differential diagnosis. Optimal immunoglobulin replacement therapy, early initiation of antibiotics, and airway clearance are essential in the management of patients with antibody deficiencies who develop pulmonary complications. Immunosuppressants may be considered in GLILD patients, especially in those with worsening pulmonary symptoms or function. In patients with bronchiectasis, low-dose azithromycin, and nebulized antibiotics have been shown to be helpful.
Patients with phagocyte disorder, namely CGD, are also at risk of developing non-infectious pulmonary manifestations from immune dysregulation. Annual chest x-rays, pulmonary function tests, and thoracic CT every 2 years, are suggested in adult CGD patients. However, there are no established guidelines for children. Apart from pulmonary infections, granulomas and malignancies should also be considered in the differential for pulmonary complications in patients with CGD. Thoracic CT, bronchoscopy, and lung biopsy should be discussed in order to distinguish these conditions from infections. A short pulse of corticosteroid is suggested as an initial treatment for granulomas in CGD patients. Other immunomodulators can be added in steroid-refractory patients with caution, and antibacterial and antifungal treatment should be continued throughout the active course of the disease.