
Abstract
Human immunodeficiency virus 1 (HIV-1) is the causative agent of AIDS. There are currently more than 35 million people living with HIV infection worldwide, and more than 2 million new infections occur each year. The global pandemic caused by HIV-1 is the subject of numerous research projects, with the development of a prophylactic vaccine and a therapeutic cure being the ultimate goals. The classic paradigms of vaccinology have proven incapable of producing a viable vaccine due to the complexity of the virus’ replication cycle, its genetic diversity, and a lack of understanding of the immune correlates of protection. Here, we briefly discuss recent vaccine approaches and the immune correlates of protection from HIV-1 infection with a focus on the role of the germinal center as a reservoir of replication-competent virus and its role in the development of broadly neutralizing antibodies in response to vaccination.
Introduction
AIDS first surfaced in the human population as an outbreak of Pneumocystis carinii pneumonia among homosexual men in the United States.1-3 It is thought to have been circulating undiscovered in Africa for many years prior. The causative agent is a Baltimore class VI retrovirus, called human immunodeficiency virus 1 (HIV-1). The virus is spread by direct sexual contact, through intravenous delivery of blood or blood products, and through vertical transmission from mother to child. The primary mode of transmission is sexual and infection is typically initiated at mucosal sites (Figure 1). As the virus replicates within host cells, it inserts a copy of its genome into that of the host—a copy that is reproduced every time that cell divides and is capable of reactivation at any time—a phenomenon called latency. The World Health Organization estimates that 75 million people have been infected since the summer of 1981, and approximately 36 million lives have been lost to the disease. Today, 0.8% of all adults aged between 15 and 50 years are living with HIV.
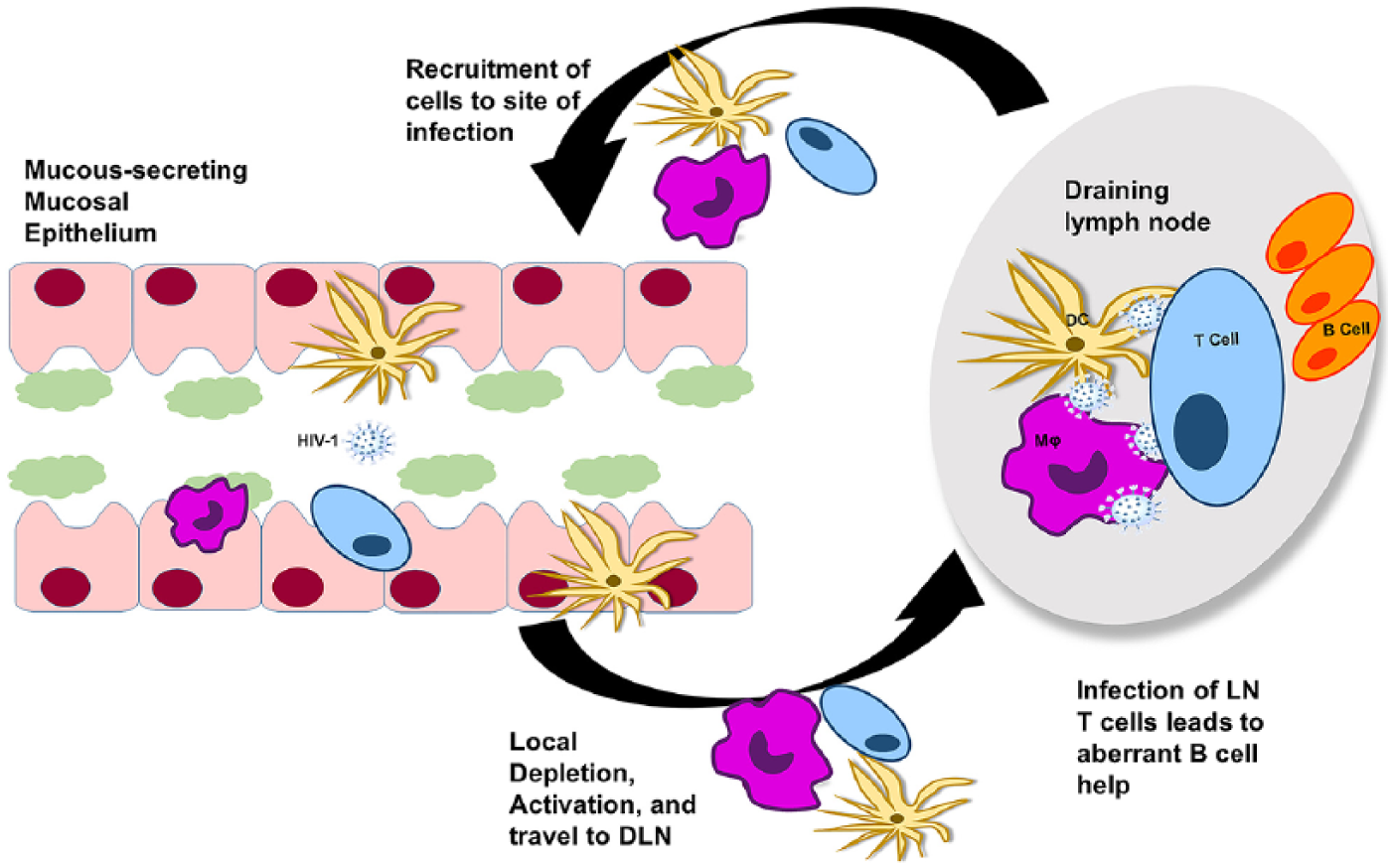
Mucosal pathogenesis of human immunodeficiency virus 1 (HIV-1). Most of the HIV-1 infections occur at mucosal sites via sexual transmission. On gaining access to tissue-resident CD4+ T cells or when taken up by tissue-resident dendritic cells (DC), infected cells may be phagocytosed by granulocytes, such as macrophages (Mϕ), and the virus is carried to the draining lymph nodes (DLNs) where these antigen-presenting cells may directly infect CD4+ T cells. Replication-competent viruses multiply and establish latency. In this way, the DLNs become the largest tissue reservoir during chronic infection.
The alarming statistics of the HIV-1 pandemic have spurred an unprecedented amount of research. Molecular medicine has provided clinicians with an arsenal of drugs that target important aspects of the viral lifecycle in hopes to decrease viral load and transmissibility. However, there is still no vaccine to prevent transmission and no feasible method to cure an HIV-1–infected individual; thus, preventing infection and removing latent virus are major research goals.
Recent discoveries have shed light on the nature of the infection and produced clues that could have major implications for a cure. In 2009, the transplant of stem cells lacking the HIV-1 co-receptor, chemokine receptor 5 (CCR5), into an HIV-1–positive patient resulted in a decrease in HIV-1 RNA to undetectable levels. 4 Recently, researchers at Temple University reported the first successful ablation of the HIV-1 proviral genome from latently infected human cells. 5 Both these represent huge advancements in HIV research and should be pursued, but at present, there are no feasible treatments for HIV-1–infected patients in places such as sub-Saharan Africa where rates of infection are the highest.
Unfortunately, all recent vaccine endeavors have proven unsuccessful. However, these failures have led to clues about the necessary correlates of protection (Table 1). Passive transfer studies have shown that neutralizing antibodies are capable of controlling HIV-1 infection and preventing infection,6-8 even though no trials to date have produced neutralizing antibodies.9-13 These results are disheartening, and solving the “neutralizing antibody problem” is the subject of much research. Adenovirus vector–delivered vaccines have not protected macaques from autologous challenge but have decreased viral loads and protected T lymphocytes, indicating that cell-mediated control of infection is also possible. The recent failures of AIDSVAX 11 and Merck Ad5 13 vaccines are disappointing but further our understanding of how to effectively prevent infection. It is clear that “traditional” vaccinology is unlikely to yield a successful therapeutic or prophylactic vaccine against HIV-1. Here, we discuss the development of anti–HIV-neutralizing antibodies, the germinal center (GC) response to HIV-1 infection, and the GC response to HIV-1 prophylactic vaccines.
Summary of past HIV-1 vaccine clinical trials.

Abbreviations: HIV-1, human immunodeficiency virus 1; NIAID, National Institute of Allergy and Infectious Diseases; NIH, National Institutes of Health.
B Cells and Neutralizing Antibodies
B lymphocytes bind antigen via a membrane-bound B-cell receptor (BCR), and they also secrete soluble forms of the BCR that can bind antigen, called antibodies. The envelope (env) protein of HIV-1 is absolutely critical for viral entry and subsequent replication; thus, antibodies that prevent env-mediated binding and entry are protective. When it was first evidenced that serum samples from some elite HIV-1 controlling patients not undergoing antiretroviral treatment could neutralize a spectrum of HIV-1 viruses,14-16 researchers grew optimistic that similar broadly neutralizing antibody (bNAb) responses could be elicited by vaccination. In fact, experiments with nonhuman primates (NHPs), as well as analyses from human elite controllers or elite neutralizers, indicate that the presence of neutralizing antibodies correlates with protection from infection.17-19 Unfortunately, these antibody responses are not typical. In general, these antibodies target conserved but less-immunogenic epitopes of the env protein or the CD4-binding site (CD4bs), whereas most of the natural responses target less well-conserved epitopes. Broadly neutralizing antibodies also tend to share unique features such as polyreactivity, a large variable heavy-chain complementarity-determining region (HCDR3), and evidence of large amounts of somatic hypermutation. In a cross-sectional study of serum samples from more than 200 patients, Hraber et al 20 found that most serum displayed some degree of cross-neutralization and that half of the samples were broadly neutralizing, but with titers at or below the thresholds thought to be protective in NHPs. 21 This indicates that most chronically infected patients develop some form of neutralizing antibody but that bNAbs are less common, and thus, although it may be feasible to induce production of neutralizing antibodies, production of antibodies that are cross-reactive but have a more restricted breadth is likely to be more feasible. Some neutralizing epitopes, such as those in the membrane-proximal external region (MPER), or the variable loops on gp41 remain hidden during infection and are generally poorly immunogenic; thus, finding ways to expose these epitopes is critical to the development of protein antigens that will incite the production of neutralizing antibodies. Novel virucides, such as the peptide triazole-thiols that cause lysis of the HIV-1 virion, present the possibility of creating novel antigens for immunization.22-24 The development of stable, soluble trimeric forms of env with native-like confirmations 25 has renewed interest in the generation of neutralizing antibody with protein vaccination. 26
When designing an immunogen intended to produce neutralizing antibody, it is important to first consider B-cell ontology. B cells develop in the bone marrow and must pass a tolerance checkpoint intended to prevent self-reactive B cells from entering peripheral circulation. It is estimated that approximately 70% of the B cells produced in the bone marrow die there due to self-reactivity. 27 Mature B cells that survive in the periphery are still not completely self-tolerant, and it is estimated that nearly 20% of these are self-reactive. 28 What this means for the development of vaccine immunogens is that the repertoire of BCRs that could respond to an immunogen is much smaller than the total number of BCRs that could be generated from the germ line. If tolerance reduces the affinity of the BCR or the number of available BCRs, this inhibits the ability of the host to mount an adequate humoral response. bNAb 2F5, which binds to the MPER of gp41, is a prime example of an atypical HIV-neutralizing antibody. 2F5 knock-in mice carrying the human heavy-chain variable (V), diversity (D), and joining (J) regions of human 2F5 29 have normal pre–B-cell development but are lacking in their ability to produce immature B cells, suggesting tolerance-induced killing of these cells at this stage of B-cell development. 30 This was later verified by the discovery that the MPER epitope bound by 2F5 (ELDKWA) is present in both the mouse and the human kynureninase enzyme.31,32
With B-cell ontology in mind, research has shifted to developing immunogens that will induce a particular B-cell lineage. When a naïve B cell encounters an antigen, it recognizes it via the BCR but also undergoes affinity maturation and somatic hypermutation as the humoral response continues to evolve such that the antibodies isolated later in an infection have much higher affinity for their antigen, that is to say, the quality of the B-cell response increases with increasing exposure to the antigen. It is also important to note that HIV rapidly mutates to escape immune pressures encountered in the host such that a constant arms-race occurs inside an infected patient, and usually the B-cell response is outpaced by viral mutagenesis. This is the reason why bNAbs are typically isolated after years of chronic infection. 33 Awareness of this phenomenon has led to the idea of designing a “reverse antigen.” This approach involves isolating a set of clonally related B cells that neutralize HIV-1 and using computational approaches to determine the original or intermediate antibodies that the neutralizing antibodies would have likely developed from during maturation of the humoral response, and designing an antigen that will be recognized by these early antibodies, thus allowing the immune system to mature until neutralizing antibodies are produced. 34 Jardine and colleagues 35 used this approach to design a gp120 mimetic that was capable of stimulating germ line BCRs and was indeed recognized by the VRC01 class of patient-isolated bNAbs, validating the approach. It has recently been demonstrated that multiple B-cell lineages may cooperate to produce bNAbs, 36 indicating that a multipronged approach to this technique is likely necessary. Similarly, in a landmark study, Wu and colleagues demonstrated that over a 15-year period, the mutational rate of antibodies in long-lived plasma cell lineages correlates with the mutational rate of HIV-1 quasispecies and that this high mutational rate leads to the development of the extraordinary antibodies isolated from latently infected patients. 37 Finally, McGuire and colleagues developed a modified env immunogen in which certain variable regions of the protein have been removed and demonstrated that this modified env was capable of eliciting germ line–reverted bNAbs as opposed to nonneutralizing antibodies. 38 These studies indicate that rational immunogen design might be used to generate HIV-1 immunogens that specifically result in the production of neutralizing antibodies.
Neutralization is not the only mechanism of antibody-mediated protection (Figure 2). Antibodies also function to induce antibody-dependent cell-mediated cytotoxicity (ADCC). Natural killer (NK) cells mediate classical ADCC by binding to the Fc region of an antibody bound to a cell that expresses viral proteins on its surface. Engagement of the NK-cell Fc receptor leads to release of cytotoxic cytokines that induce apoptosis in the target cell. Studies have shown that early ADCC responses to HIV env proteins can control viremia. 39 Interestingly, it has also been shown that the specificity of the antibodies mediating ADCC differs between HIV controllers and noncontrollers, whereas the functionality of their NK cells remains the same with HIV controllers more often producing env-targeted antibodies. 40 Antibodies can also mediate phagocytosis, complement activation, and mucus entrapment.41-43 In attempts to cure chronic HIV-1 infection, nonclassical HIV immunogens, such as those targeting the transactivator (Tat), have been developed. Indeed, it has been shown that antibodies targeting Tat are capable of preventing HIV reactivation and limiting pathogenesis. 44

Role of antibodies in HIV-1 infection. Affinity-matured, class-switched, highly-mutated, long-lived plasma cells can secrete anti–HIV-neutralizing antibodies that are capable of neutralizing free virus to prevent infection of target cells (left) as well as mediating ADCC or ADCP (middle) by natural killer cells. Quiescent cells that are latently infected remain immune to anti-HIV responses (right). ADCC indicates antibody-dependent cell-mediated cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; HIV-1, human immunodeficiency virus 1; NK, natural killer.
Although production of bNAb is important to prevent primary HIV-1 infection, it is important to note that the quality of antibody produced as a result of infection or vaccination is greatly enhanced by optimal help from the cell-mediated arm of the immune system. B cells that are activated independently of CD4+ helper T cells produce polyclonal antibodies. However, GC formation and the processes of affinity maturation and isotype switching of antibodies can only occur with the help of T cells. The goal of vaccination would be to prime recipients and induce a memory response that is protective against subsequent HIV-1 challenge, but memory B cells are formed only in GCs. For this reason, it is unlikely that a vaccine platform that solely elicits a B-cell response will provide long-lived immunity. Protein antigens are highly immunogenic and elicit primarily an antibody-mediated response, which may help neutralize infectious viral particles initially but will likely not lead to a robust recall response; thus, a combination of platforms is a better option.
With current data indicating that the traditional approach to vaccine development is likely to be ineffective against HIV, researchers have developed unique antigens and antigen delivery systems. The use of adenovirus and adeno-associated virus (AAV) serotypes to deliver HIV antigens proved a promising way to achieve long-lasting expression of antigens45-47 but can be hindered by the presence of preexisting host immunity against the vector.48,49 As the most conserved regions of the envelope protein are the CD4- and co-receptor-binding sites, recombinant fusion proteins targeting these epitopes provide a nontraditional means of controlling or preventing HIV infections.50-52 Interestingly, AAV vectors have been used as a gene therapy vehicle to deliver these molecules. 50 In the most promising of these approaches, the coding sequence of eCD4-Ig, a recombinant immunoadhesin form of CD4 (CD4-Ig), and CCR5mim1, a CCR5 mimetic peptide derived from bNAb E51, were delivered to rhesus macaque via intramuscular injection of AAV. Gardner and colleagues demonstrate that eCD4-Ig bound and neutralized most of the HIV-1 isolates better than either of the components alone and was also capable of neutralizing simian/human immunodeficiency virus (SHIV) isolates. The group also demonstrated stable expression of the construct up to 40 weeks post inoculation in rhesus macaques and protection against challenge with a variety of SHIVs. 53 Other promising vectors include modified cytomegalovirus 54 and adenovirus serotype 26 (Ad26) which may avoid the issue of preexisting immunity that hinders other serotypes. 55
The GC in HIV-1 Infection
It is important to note that the germinal center reaction (GCR) occurs in the presence of HIV-1 virions. Dendritic cells are known to carry immune complexes containing HIV-1 virions on their processes to the GC, and follicular helper T (TFH) cells are CD4+ and thus targets for HIV-1 infection. Despite this, HIV-1–infected patients have higher numbers of TFH cells and GC B cells,56,57 leading to increased numbers of circulating B cells, hypergammaglobulinemia, and, in some cases, B-cell lymphomas. 58 It is well established that CD4+ T cells in the lymph nodes serve as reservoirs for HIV-1.57,59,60 Recently, Kohler et al 61 used spinoculation of human tonsillar cells to demonstrate that GC TFH are highly permissive to HIV infection, more so than extrafollicular or non-GC TFH which express low levels of programmed death 1 (PD-1) and C-X-C chemokine receptor 5 (CXCR5). They further demonstrated, by in situ hybridization, the presence of increased HIV-1 RNA in the GCs compared with the extrafollicular areas in sections of inguinal lymph node biopsies from HIV-1–positive, treatment-naïve, non-AIDS patients. It is also well established that TFH from HIV-positive patients provide inadequate help to B cells in vitro, 62 but the underlying cause of this impairment has not been definitively determined. Using a unique cohort of organ transplant patients or patients requiring splenectomy, Colineau et al attempted to elucidate the underlying causes of this impairment. The group determined that there was an increase in GC TFH in HIV-positive patients but that these cells had decreased messenger RNA (mRNA) expression of CD40L, OX40, and inducible costimulator (ICOS)—molecules involved in TFH stimulation. 63 Interestingly, although these cells had decreased mRNA levels of STAT3, a master signal transducer required for maintenance of the TFH phenotype, they did not have decreased Bcl6 expression compared with HIV-1–negative cells. Despite this, the number of follicular regulatory T (TFR) cells in HIV-1–positive spleens increased, indicative of continued GC reactions which correlate with the increased numbers of TFH and B cells observed in HIV patients. 63 Understanding the source of the defect in TFH help during HIV-1 infection is imperative to the development of an HIV cure.
Although it is evident that lymph node GC TFH cells are major reservoirs of HIV-1 virus in chronically infected patients, 59 it appears that this location is somehow protected from CD8+ T-cell immune surveillance. There is evidence that the acute increase in CD8+ T cells coincides with a decrease in viremia, 64 supporting the notion that CD8+ T-cell responses can mediate clearance of infected cells. Access to lymph node GCs is dependent on the CXCR5/CXCL13 axis. CXCR5+ CD8+ T cells have been shown to localize to the B-cell follicle and even exhibit a regulatory function, at least in the context of B-cell lymphoma. 65 Recently, CXCR5+CD44hiCD8+ T cells with regulatory activity (secretion of interleukin [IL]-10) have been demonstrated to limit HIV replication in TFH, impair IL-21 production, and inhibit IgG secretion during ex vivo HIV infection, indicating a unique CD8+ regulatory component of GCs during HIV infection. 66 Further studies are needed to characterize the access of CD8+ T cells to the GC and to evaluate whether these cells could kill HIV-1–infected TFH cells therein.
The GC and Vaccine-Induced Responses
Germinal centers are dynamic sites within lymphoid organs where mature B cells undergo somatic hypermutation, affinity maturation, and class-switch recombination of the BCR. The outcome of the GCR is that these mature B cells differentiate into long-lived plasma cells and memory B cells, the cells responsible for instituting humoral immunologic memory (Figure 3). Humoral memory is the hallmark of successful vaccination and memory B cells mediate this memory; therefore, optimizing the GC response to HIV-1 vaccine antigens is integral to the development of effective HIV prophylactics.

The germinal center reaction. On antigen presentation in the context of the proper cytokine milieu, naïve CD4+ T cells differentiate into follicular helper T (TFH) cells and migrate to germinal centers where they select germinal center B cells (GC B) which are responsive to the appropriate antigen via presentation of the antigen on major histocompatibility complex (MHC I). Both TFH and GC B cells traffic to the germinal center by virtue of their expression of the receptor CXCR5, following a chemokine gradient of the CXCR5 ligand CXCL13. TFH cells and GC B cells make physical connection between costimulatory molecules, B7 and CD28, CD40 and CD40L, inducible costimulator (ICOS) and ICOS ligand (ICOSL), as well as PD-1 and PD-1L. This interaction is also cytokine based with the TFH cell secreting interleukin (IL)-21 which is recognized by the IL-12R on GC B cells and mediates STAT3 support of affinity maturation, somatic hypermutation, and class switching of antibodies. TFH cells also secrete IL-4 which is recognized by the IL-4 receptor on GC B cells and supports STAT3 expression. Antigen-specific dendritic cells which also present antigen to TFH secrete IL-6 which promotes Bcl-6 expression in TFH and GC B supporting the GC phenotype. Finally, this reaction is subject to regulation by T follicular regulatory (TFR) cells which secrete IL-10 and inhibit both TFH and GC B function.
The selection and differentiation of B cells in the GC are mediated by a special subset of CD4+ T cells called TFH cells. Follicular helper T cells differentiate from naïve CD4+ T cells when antigen is presented to them by antigen-presenting cells that secrete the necessary cytokine milieu, namely, the cytokines IL-21, 67 IL-6, 68 and IL-12. 69 After acquiring the TFH phenotype, these cells migrate to the GC by virtue of their expression of CXCR5 following a gradient of the ligand for CXCR5, C-X-C ligand 13 (CXCL13).70,71 In the GC, TFH cells interact both physically and via cytokine signaling with GC B cells. The physical interaction between GC B cells and TFH includes ICOS on TFH and its ligand ICOSL, PD-1 72 and its ligand PD-L1, 73 CD80/CD86, 74 and CD40/CD40L. 75 Follicular helper T cells signal GC B-cell differentiation into memory B cells and long-lived plasma cells by secreting IL-4 and IL-21.76-78 Follicular helper T cells are typically identified by the expression of CXCR5, CD4, PD-1, and the transcription factor Bcl-6. It has been shown that PD-1 signaling through PD-L1 is essential for the development of plasma cells and that deficiency in PD-1 signaling leads to a decrease in the number of long-lived plasma cells. 79
As the GCR is critical to the development of humoral immunity and immune memory, studying this response is critical to the development of vaccines targeting pathogens. Unfortunately, direct examination of the GC response in human patients is rarely feasible. CD4+ cells possessing TFH-like phenotype (CXCR5+ PD-1+) cells can be isolated from the periphery, and it has been demonstrated that an early preservation of B and T cells with this phenotype predicts a broad neutralizing antibody response in HIV-1 chronically infected patients.80,81 These peripheral TFH-like cells have also been connected to the ability to develop an effective humoral response to vaccination. 82 He et al 83 reported that a population of CCR7loPD-1HiCXCR+ T cells correlated with the formation of GCs in both humans and mice, whereas a CCR7HiPD-1LoCXCR5+ CD4+ T cell possessed a resting, memory-like phenotype and could not be used as a biomarker for GC activity. In another study, conflicting results were observed; when measuring CCR7HiCXCR5+PD-1Hi cells in the periphery TFH (pTFH) cells, Boswell et al 84 report no relationship between serum-neutralizing capability and pTFH cells, env-specific antibody, or total plasma immunoglobulin levels even though these cells were capable of in vitro B-cell help, concluding that these cells possess a memory-like phenotype as opposed to a classical GC TFH phenotype. These conflicting results indicate that another measure of the GCR is necessary to gauge vaccine-induced responses. Recently, Havenar-Daughton et al 85 reported the use of soluble CXCL13, the chemokine that attracts TFH and GC B cells to lymph node GCs, in blood as a biomarker for GC activity. The group reported that elevated CXCL13 levels correlated with the development of anti–HIV-1 bNAbs in the well-characterized International AIDS Vaccine Initiative protocol C cohort,82,86 as well as in immunized mice and macaques. 85 Measurement of soluble CXCL13 could provide a feasible means of evaluating HIV-1 vaccine–induced responses as well as a clinical correlate of HIV infection in the lymph node. Recently, this same group correlated the development of tier 2 HIV-neutralizing antibodies post env trimer immunization with GC B-cell and TFH-cell frequencies in a rhesus macaque model using longitudinal fine-needle aspiration. 87
Finally, inciting a robust GC response to vaccine antigens would mean seeding an antigen-experienced memory cell pool. As TFH select B cells with the appropriate affinity and support their differentiation into long-lived plasma cells and memory B cells, a vaccine that promotes the induction of TFH cells and GCs is likely to induce immunologic memory with fewer immunizations. Although the precise kinetics of memory B-cell formation post antigen exposure are under debate, 88 it is still widely held that the generation of memory B and plasma cells following exposure to T-dependent antigens depends on the GCR. The TFH phenotype is promoted by signaling through ILs 12, 6, and 21. It has recently been demonstrated that the oil-in-water formulated adjuvant MF59, which has been shown to induce broad, potent, protective humoral responses to influenza vaccination, mediates its adjuvant effect by enhancing TFH cells in early and adult life.89,90 Interestingly, it was also reported that this TFH enhancement observed in young adult and adult mice is absent in 1-week-old neonatal mice due to an increased number of TFR cells which suppress the GCR. 89 A recent study undertaken to evaluate the benefit of using MF59 instead of alum in the ALVAC vaccine formulation discovered that although MF59 has been shown to enhance GC formation, it did not lead to increased protection from challenge in an SHIV model. 91 The plasticity of CD4+ T cells provides another potential avenue for augmenting the GC response. It has been shown that TH17 cells preferentially differentiate into TFH cells in the mucosa and that this plasticity is causally linked to IgA production in the gut.92,93 IgA production has been linked to protection against HIV-1 infection.94-96 This could mean that TH17 cells are an, as yet, unexplored means of enhancing the GC response and increasing vaccine-induced IgA production in the mucosa, which is the primary site of HIV-1 transmission. DNA vaccines incorporating plasmid-encoded IL-12 have demonstrated the ability to enhance humoral responses to HIV-1 DNA vaccines.97-99 This humoral enhancement is partially linked to increased GC responses induced when IL-12 is included as a molecular cytokine adjuvant (Gary and Kutzler, unpublished results). Interleukin 12 is thought to mediate this effect via STAT4 which supports Bcl-6 expression which promotes and maintains the TFH and GC B phenotype. 69 Notably, a defect in IL-12 signaling interferes with TFH cell function in vivo. 100 The delivery of malaria antigens formulated with nanoparticle-based delivery was recently demonstrated to enhance GC formation and TFH expansion compared with the adjuvant monophosphoryl lipid A. 101 The inclusion of ligands that bind the retinoic acid–inducible gene-I pattern recognition receptor pathway, such as synthetic double-stranded RNA (dsRNA), has also been shown to enhance the GC response to influenza antigens. 102 Adenosine deaminase, a member of the growth factor cytokine family, has been demonstrated to enhance the costimulatory effect of DCs on CD4+ T cells 103 and has been shown to enhance T-cell responses elicited by DCs loaded with inactivated HIV viruses. 104
The discovery of adjuvants or vaccine platforms that enhance the GC response is integral to the development of vaccines with shorter immunization courses (Table 2). Inciting a robust GC response and seeding the memory B-cell pool following a single immunization will mean fewer immunizations, leading to dose sparing and reduced production costs, all important for delivering vaccines to the developing world. The development of reliable biomarkers to gauge the GCR will be critical to evaluate the efficacy of vaccines and adjuvants targeting this reaction.
Summary of vaccine adjuvants targeting the germinal center reaction.
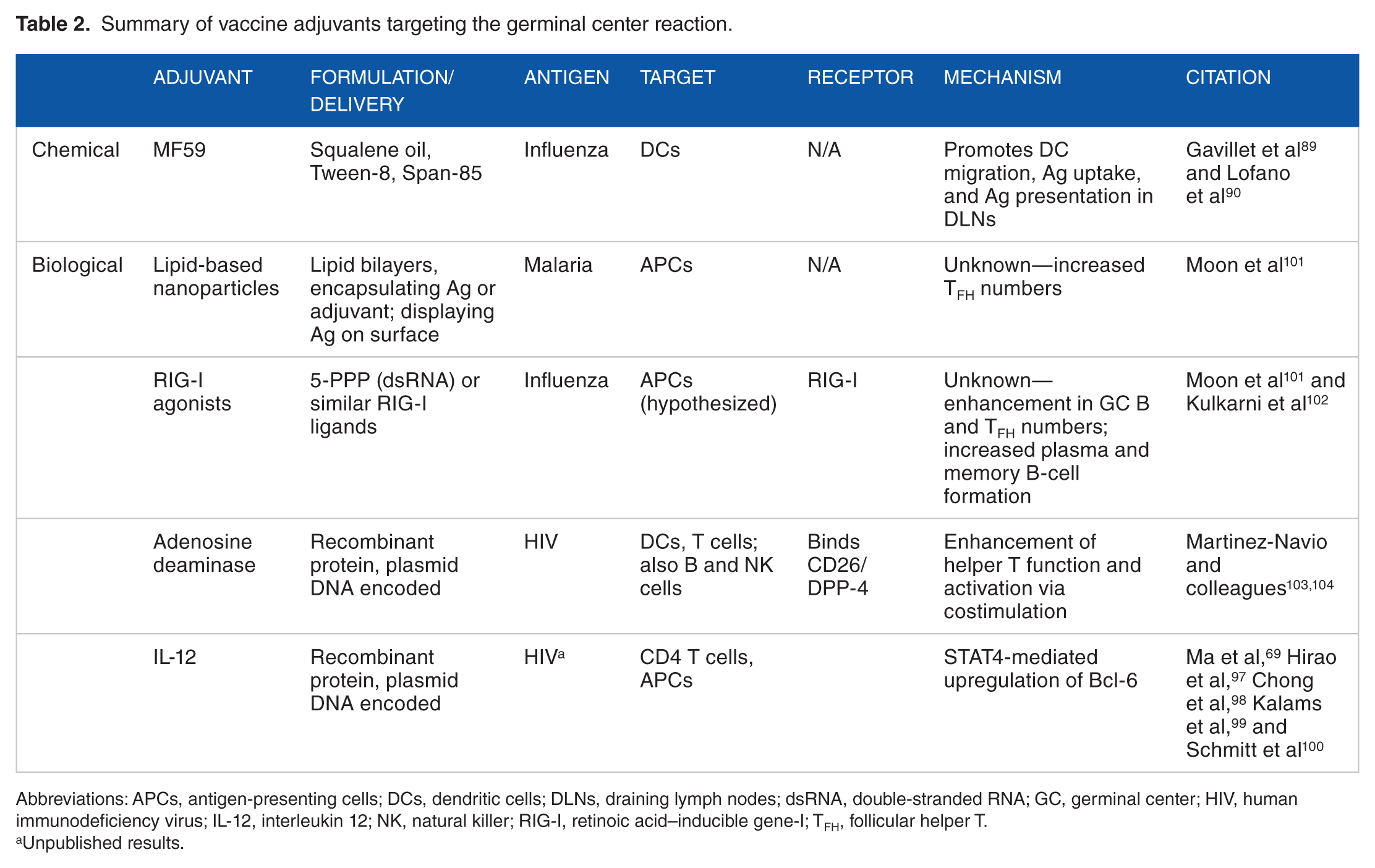
Abbreviations: APCs, antigen-presenting cells; DCs, dendritic cells; DLNs, draining lymph nodes; dsRNA, double-stranded RNA; GC, germinal center; HIV, human immunodeficiency virus; IL-12, interleukin 12; NK, natural killer; RIG-I, retinoic acid–inducible gene-I; TFH, follicular helper T.
Unpublished results.
Perspectives and Future Challenges
The AIDS epidemic has been raging for more than 3 decades. Great advancements have been made in treatment, and the life expectancy of an HIV-1–infected individual approaches that of uninfected individuals. Our ability to drastically decrease the morbidity and mortality associated with chronic infection is impressive; however, 2.5 million people worldwide become infected with HIV-1 each year, that is, more than 6000 people every day. These intimidating statistics inspire continued research aimed at the development of a prophylactic vaccine.
As immunologic knowledge improves, the ability to create more effective prophylactics increases. In the past 30 years, more has been learned about the types of immune responses elicited by different vaccine platforms since the advent of vaccinology, and this is due in great part to HIV-1 research. Despite this, several obstacles still impede us—preexisting immunity to HIV vaccine vectors may inhibit the efficacy of vaccine candidates, adequate animal models to accurately predict protection in humans do not exist, and the evolution of the virus in response to host selection pressure cannot be accurately predicted. The following question remains: how do we develop a prophylactic vaccine against HIV-1 that is highly immunogenic in humans? The vaccine needs to elicit a neutralizing antibody response, with high titers of plasma antibodies, as well as a large pool of memory B cells, but this response requires TFH cells that are themselves permissive to infection.
The formulation of such a vaccine remains elusive. Protein-based vaccines generally evoke strong antibody responses, and live vectored vaccines can initiate both arms of the immune response, provided they are not hindered by preexisting immunity. DNA vaccines elicit strong cell-mediated responses and appear capable of engendering an effective TFH response, but the recent DNA prime-adenovirus boost human trial did not demonstrate efficacy. 13 The only approach to demonstrate efficacy to date was the initial phase 2 trial using a canary pox vector bearing HIV-1 gag, pol, and env proteins, boosted with a recombinant gp120 protein, and still the vaccine proved only 25% effective at preventing infection. The plasticity of CD4+ T-cell subsets is of particular interest. Recent studies have demonstrated the ability of mucosal resident TH17 cells to differentiate into GC TFH cells and stimulate mucosal antibody secretion. It may be feasible to target these cells prior to differentiation via vaccination, priming an exceptional GCR.
The failure of previous vaccine trial has revealed the complex nature of a successful immune response against HIV-1 as well as the types of immune responses engendered by the different platforms available. This makes better assumptions about the outcomes of a trial possible. The prime-boost regimen will likely prove to be the most effective method for prompting both a humoral and a cell-mediated response. This dearth of knowledge has led to creative approaches to solving the HIV-1 pandemic.
Footnotes
Peer review:
Six peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1179 words, excluding any confidential comments to the academic editor.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was partially funded under the Ruth L. Kirschstein National Research Service Award 5T32MH079785 (Rappaport, principal investigator, and Wigdahl, principal investigator of subcontract to Drexel University College of Medicine) as E.G. is a trainee on this grant. The contents of the paper are solely the responsibility of the authors and do not necessarily represent the official views of the national Institutes of Health. This work is also supported by the W.W. Smith Charitable Trust Foundation grant A1404 to M.K.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
ENG conceptualized and wrote this article with comments and guidance from MAK.