
Abstract
Background:
The bevacizumab biosimilar (Encoda), which was approved by the National Medical Products Administration (NMPA) in China in 2019, is a biosimilar of bevacizumab. Approval of bevacizumab biosimilar (Encoda) for metastatic colorectal cancer (mCRC) was based on the extrapolation principle of biosimilar. However, there is currently no available data regarding the efficacy and safety of both bevacizumab biosimilar (Encoda) and bevacizumab in patients with mCRC.
Methods:
The present real-world study included patients with mCRC who received first-line therapy with either bevacizumab biosimilar (Encoda) or bevacizumab combined with backbone chemotherapy between April 2021 and December 2022. The overall response rate (ORR) was the primary endpoint of the study. Bevacizumab biosimilar (Encoda) would be considered equivalent to bevacizumab if it met any of the following criteria: the 90% CI for ORR risk ratio of bevacizumab biosimilar (Encoda): bevacizumab was included within the predefined equivalence range (0.75 to 1.33) as specified by the NMPA or within the predefined equivalence range (0.73 to 1.36) as specified by US Food and Drug Administration (FDA); the 95% CI for the ORR risk difference of 2 groups was included within the predefined equivalence range (−0.12 to 0.15) as specified by the European Medicines Agency (EMA).
Results:
The study included a total of 436 patients, with 234 receiving bevacizumab biosimilar (Encoda) and 202 receiving bevacizumab. The ORR was 42.3% (95% CI: 35.9% to 48.9%) in the bevacizumab biosimilar (Encoda) group and 42.1% (95% CI: 35.2% to 49.2%) in the bevacizumab group. The ORR risk ratio and risk difference were 1.005 (90% CI: 0.836 to 1.210) and 0.002 (95% CI: −0.091 to 0.095), respectively, both included within the prespecified equivalence range. The overall safety profile was comparable between the 2 groups.
Conclusions:
This real-world study is the first to demonstrate the efficacy equivalence of bevacizumab biosimilar (Encoda) and bevacizumab in first-line treatment of mCRC.
Introduction
Globally, colorectal cancer (CRC) exhibits the highest incidence and fatality rates, ranking second and third, respectively. Colorectal cancer accounts for approximately 10% of all cancer diagnoses and cancer-related deaths annually. 1 Most patients diagnosed with CRC will ultimately develop metastatic colorectal cancer (mCRC). 2 In recent years, novel anti-tumor drugs targeting vascular endothelial growth factor (VEGF) or epidermal growth factor receptors have significantly contributed to the treatment of mCRC. 3 Bevacizumab (Avastin) is a recombinant humanized monoclonal antibody that specifically binds to VEGF, thereby preventing the interaction between VEGF and its receptors on the surface of endothelial cells. This inhibition leads to reduced VEGF bioactivity and subsequent suppression of microvascular growth. 4 The combination of bevacizumab and backbone chemotherapy has gained approval for the treatment of mCRC in numerous countries worldwide, and is recommended as the first-line standard of care for mCRC by esteemed organizations such as the National Comprehensive Cancer Network, the European Society for Medical Oncology, and National Health Commission of the People’s Republic of China. 3
The second bevacizumab biosimilar, Encoda (Qilu Pharmaceutical Group Co, Ltd.), received approval from the National Medical Products Administration (NMPA) in China in 2019. 5 The relatively low cost of biosimilar research and development results in lower pricing compared with reference biologics, offering more affordable treatment options for patients. In addition, the introduction of biosimilars creates intense market competition, prompting reference biologics to reduce prices to maintain their market share, ultimately leading to a reduction in health care expenditure. These biosimilars are estimated to have saved the EU $11.8 billion between 2016 and 2022. 6 The current price of bevacizumab biosimilar (Encoda) in China is 25% lower than that of bevacizumab, and the extensive utilization of bevacizumab biosimilar (Encoda) is expected to alleviate the economic burden on patients. Prospective clinical trials have demonstrated equivalent efficacy between bevacizumab and its biosimilar (Encoda) in the treatment of nonsquamous non-small cell lung cancer (NSCLC).5,7 According to the current policy, indication extrapolation is based on the overall similarity between the proposed biosimilar and the reference product. When clinical trials can demonstrate that the proposed biosimilar is clinically equivalent to the reference product in at least one indication, scientific evidence from research data and information related to extrapolated indications may support direct utilization of the proposed biosimilar for other indications approved for use by the reference product. 8 The approval of bevacizumab biosimilar (Encoda) for the treatment of mCRC was based on the principle of indication extrapolation rather than clinical trial validation. However, unanswered questions persist regarding the comparability of the effectiveness and safety between bevacizumab biosimilar (Encoda) and the reference product in patients with mCRC. The utilization of bevacizumab biosimilar (Encoda) in real-world scenarios for the treatment of mCRC will address this research gap and establish a crucial foundation for therapeutic drug selection.
The present real-world study presents the first-ever efficacy and safety data comparing bevacizumab biosimilar (Encoda) to the reference bevacizumab in the treatment of mCRC. The primary objective of this study was to establish the equivalence between bevacizumab biosimilar (Encoda) combined with backbone chemotherapy and bevacizumab combined with backbone chemotherapy as first-line treatment for mCRC. The safety profiles of bevacizumab biosimilar (Encoda) and bevacizumab in patients with mCRC were compared, additionally.
Patients and Methods
Patients and study design
The purpose of this multicenter, real-world, observational study was to compare the efficacy and safety profiles of bevacizumab biosimilar (Encoda) with reference bevacizumab. The patients included in this study were from 2 institutions in China, namely Fudan University Shanghai Cancer Center and Zhongshan Hospital Fudan University. The inclusion criteria comprised the following: (1) histologically and pathologically proven, unresectable mCRC; (2) Eastern Cooperative Oncology Group performance status (ECOG PS) 9 score ⩽ 2; (3) received backbone chemotherapy combined with bevacizumab biosimilar (Encoda) or bevacizumab as first-line therapy between April 2021 and December 2022; (4) backbone chemotherapy regimens including capecitabine plus oxaliplatin (XELOX), 5-fluorouracil (5-FU), leucovorin and oxaliplatin (FOLFOX), 5-FU, leucovorin and irinotecan (FOLFIRI) or 5-FU, leucovorin, oxaliplatin and irinotecan (FOLFOXIRI); and (5) dosage according to the standard dose for each protocol. Exclusion criteria consisted of (1) patients who refused to enroll in the study or refused to cooperate with follow-up and (2) patients who had participated in other intervention studies within the past 4 weeks. Data on patients were collected either prospectively or retrospectively. In December 2022, patients who were not prospectively screened and who received bevacizumab or biosimilar between April 2021 and December 2022 were retrospectively screened (Figure 1).

Flowchart of patient selection for both prospective (A) and retrospective (B) data analysis. Biosimilar, bevacizumab biosimilar (Encoda); ECOG PS, Eastern Cooperative Oncology Group performance status; FOLFIRI, 5-fluorouracil, leucovorin, and irinotecan; FOLFOX, 5-fluorouracil, leucovorin, and oxaliplatin; FOLFOXIRI, 5-fluorouracil, leucovorin, oxaliplatin and irinotecan; XELOX, capecitabine plus oxaliplatin.
Procedures
Bevacizumab biosimilar (Encoda) or bevacizumab was administered intravenously (7.5 mg/kg 21-day cycle combined with XELOX or 5 mg/kg 14-day cycle combined with FOLFOX, FOLFIRI, or FOLFOXIRI). The XELOX regimen consisted of oxaliplatin 130 mg/m2 intravenously on day 1, followed by oral capecitabine 1 g/m2 twice daily on days 1 to 14 of the 21-day cycle. The FOLFOX regimen consisted of oxaliplatin 85 mg/m2 intravenously, followed by leucovorin 400 mg/m2 intravenously, and 5-FU 400 mg/m2 bolus on day 1 with subsequent 5-FU 2.4 g/m2 × 46 hours continuous intravenous infusion, repeated every 14 days. The FOLFIRI regimen consisted of irinotecan 180 mg/m2 intravenously, followed by leucovorin 400 mg/m2 intravenously, and 5-FU 400 mg/m2 bolus on day 1 with subsequent 5-FU 2.4 g/m2 × 46 hours continuous intravenous infusion, repeated every 14 days. The FOLFOXIRI regimen consisted of oxaliplatin 85 mg/m2 intravenously, leucovorin 400 mg/m2 intravenously, irinotecan 165 mg/m2 intravenously, and 5-FU 400 mg/m2 bolus on day 1 with subsequent 5-FU 2.4 g/m2 × 46 hours continuous intravenous infusion, repeated every 14 days. The combination treatments were administered for up to 12 cycles followed by the administration of capecitabine or 5-FU/leucovorin in addition to bevacizumab biosimilar (Encoda) or bevacizumab maintenance until disease progression (PD), occurrence of intolerable adverse events, or when the patient was eligible for radical excision or radiation therapy.
Endpoints and assessments
The primary endpoint of the study was the overall response rate (ORR), which was calculated as the proportion of patients whose best response, assessed by independent and blinded central imaging review basing on Response Evaluation Criteria in Solid Tumors version 1.1, was classified as complete response (CR) or partial response (PR). The secondary endpoint was progression-free survival (PFS), defined as the time from initiation of bevacizumab biosimilar (Encoda) or bevacizumab until onset of PD, death from any cause, or final follow-up. The safety endpoint was used for the comparison of treatment-emergent adverse events (TEAEs) between bevacizumab biosimilar (Encoda) and bevacizumab. Adverse events (AEs) were assessed and classified based on the National Cancer Institute Common Terminology Criteria for Adverse Events, version 5.0.
Statistical analysis
Clinical equivalence was determined by analyzing the risk ratio and risk difference of ORR between the treatment groups. Bevacizumab biosimilar (Encoda) was deemed equivalent to bevacizumab if it met any of the following conditions: the 90% CI for ORR risk ratio of bevacizumab biosimilar (Encoda): bevacizumab was included within the predefined equivalence range (0.75 to 1.33) as specified by the NMPA or within the predefined equivalence range (0.73 to 1.36) as specified by US Food and Drug Administration (FDA); the 95% CI for the ORR risk difference of 2 groups was included within the predefined equivalence range (−0.12 to 0.15) as specified by the European Medicines Agency (EMA). 10
Fisher exact test or chi-squared test was employed for the analysis of categorical variables. The continuous variables, represented as medians, were assessed using Wilcoxon Mann-Whitney test. The log-rank test was employed to analyze the Kaplan–Meier survival curves. The factors influencing survival were assessed using a Cox proportional hazards model, and the corresponding results were presented in hazard ratios (HRs) and 95% CIs. Differences were considered statistically significant at P values of <0.05. Graphical plotting and statistical analyses were presented using GraphPad Prism software (version 5.01; Dotmatics, Boston, Massachusetts), IBM SPSS Statistics software (version 21.0; IBM Corp., Armonk, New York), and SAS software (version 9.4; SAS Institute, Cary, North Carolina).
Results
Patient and treatment characteristics
The study enrolled a total of 436 patients, with 89 (20.4%) being prospectively recruited. Among them, 234 patients received bevacizumab biosimilar (Encoda) in combination with backbone chemotherapy, while 202 patients received bevacizumab combined with backbone chemotherapy. The median follow-up time was 14.1 months (range, 1.0 to 35.9). Table 1 provides a summary of the patients’ characteristics, including age, gender, ECOG PS, chemotherapy regimen, primary tumor sidedness, metastatic sites, RAS/BRAF status, DNA mismatch-repair (MMR) status, and concomitant diseases. There were no significant differences in demographic and baseline characteristics between the 2 treatment groups (Table 1). The overall median age was 60 years, with the bevacizumab biosimilar (Encoda) group having a median age of 61 years and the bevacizumab group having a median age of 59.5 years. Out of the 436 patients, 258 (59.2%) were men, with the bevacizumab biosimilar (Encoda) group comprising 59.4% men and the bevacizumab group comprising 58.9% men. Most backbone chemotherapy regimen was XELOX (bevacizumab biosimilar [Encoda], 48.7%; bevacizumab, 46.5%), followed by FOLFIRI (bevacizumab biosimilar [Encoda], 27.4%; bevacizumab, 26.7%). Before initial treatment with bevacizumab biosimilar (Encoda) or bevacizumab, 232 patients (53.2%) had 1 site metastasis without peritoneal metastases, 128 patients (29.4%) had 2 or more sites metastases without peritoneal metastases, and 76 patients (17.4%) had peritoneal metastases.
Characteristic of patients at baseline.

Abbreviations: Biosimilar, bevacizumab biosimilar (Encoda); ECOG PS, Eastern Cooperative Oncology Group performance status; FOLFIRI, 5-fluorouracil, leucovorin, and irinotecan; FOLFOX, 5-fluorouracil, leucovorin, and oxaliplatin; FOLFOXIRI, 5-fluorouracil, leucovorin, oxaliplatin and irinotecan; MMR, DNA mismatch-repair; N/A, not available; XELOX, capecitabine plus oxaliplatin.
KRAS mutation gene accounted for the largest proportion of patients (56.9%), followed by RAS and BRAF wild-type (18.3%). The most common MMR status type was pMMR (84.6%). The most common concomitant diseases were hypertension (23.9%), diabetes (11.9%), and autoimmune disease (3.0%).
There was no significant difference in exposure to bevacizumab biosimilar (Encoda) or bevacizumab between the 2 treatment groups. The mean exposure time of bevacizumab biosimilar (Encoda) and bevacizumab was 5.974 months (standard deviation [SD], 4.857) and 5.961 months (5.195), respectively. The mean dose number of bevacizumab biosimilar (Encoda) and bevacizumab was 7.899 (5.474) and 8.298 (6.761), respectively, in the 21-day regimen and 10.067 (6.967) and 10.009 (7.345) in the 14-day regimen. The mean dose intensity of bevacizumab biosimilar (Encoda) and bevacizumab was 6.245 and 6.243 mg/kg/cycle, respectively.
Efficacy
In the bevacizumab biosimilar (Encoda) group (n = 234), 2 patients achieved CR and 97 patients achieved PR, and the ORR in this group was 42.3% (95% CI: 35.9% to 48.9%) as shown in Table 2. In the bevacizumab group (n = 202), 1 patient achieved CR and 84 patients achieved PR, and the ORR in this group was 42.1% (95% CI: 35.2% to 49.2%). The unstratified ORR risk ratio and the unstratified ORR risk difference were 1.005 (95% CI: 0.807 to 1.253, 90% CI: 0.836 to 1.210) and 0.002 (95% CI: −0.091 to 0.095, 90% CI: −0.076 to 0.080), respectively. The 90% CI of the ORR risk ratio for bevacizumab biosimilar (Encoda)/bevacizumab fell comfortably within the NMPA and FDA equivalent range, while the 95% CI of the ORR risk difference for bevacizumab biosimilar (Encoda)/bevacizumab was well within equivalent range defined by EMA. These results indicate that bevacizumab biosimilar (Encoda) and bevacizumab are clinically comparable.
Comparison of overall response rate between Encoda and bevacizumab groups.

Abbreviations: Biosimilar, bevacizumab biosimilar (Encoda); CI, confidential interval; CR, complete response; ORR, overall response rate; PR, partial response; SD, stable disease; PD, progressive disease.
The PFS analyses were conducted based on 110 events (47.0%) in the bevacizumab biosimilar (Encoda) group and 121 events (59.9%) in the bevacizumab group. There was no significant difference observed in the median PFS interval between the 2 treatment groups, with a duration of 11.2 months (95% CI: 9.01 to 13.39) for the bevacizumab biosimilar (Encoda) group and 10.4 months (95% CI: 8.89 to 11.91) for the bevacizumab group, respectively (HR = 0.87, 95% CI: 0.67 to 1.13; P = 0.426) as shown in Figure 2. The median follow-up time was 14.1 months (range, 1.0 to 35.9), surpassing the median PFS time, indicating that the PFS results were reliable. These results imply that bevacizumab biosimilar (Encoda) and bevacizumab exhibit comparable long-term efficacy.

Kaplan-Meier curves of PFS for patients who received bevacizumab biosimilar (Encoda) or bevacizumab in combination with backbone chemotherapy as first-line treatment. Biosimilar, bevacizumab biosimilar (Encoda); HR, hazard ratio; mo: months; PFS, progression-free survival.
Safety
The incidence of TEAEs among all enrolled patients was 83.7% (365/436), as indicated in Table 3. Among these, 104 cases (23.9%) were classified as grade 3 or higher AEs. There was no significant difference in the incidence of TEAEs at any grade between the bevacizumab biosimilar (Encoda) and bevacizumab groups, with frequencies of 194 cases (82.9%) and 171 cases (84.7%), respectively. Most TEAEs reported were categorized as grade 1 or 2, while the occurrence rates of grade 3 or 4 TEAEs were comparable between the bevacizumab biosimilar (Encoda) group and the bevacizumab group, with percentages of 22.6% and 25.2%, respectively .
Summary of treatment-emergent adverse events.
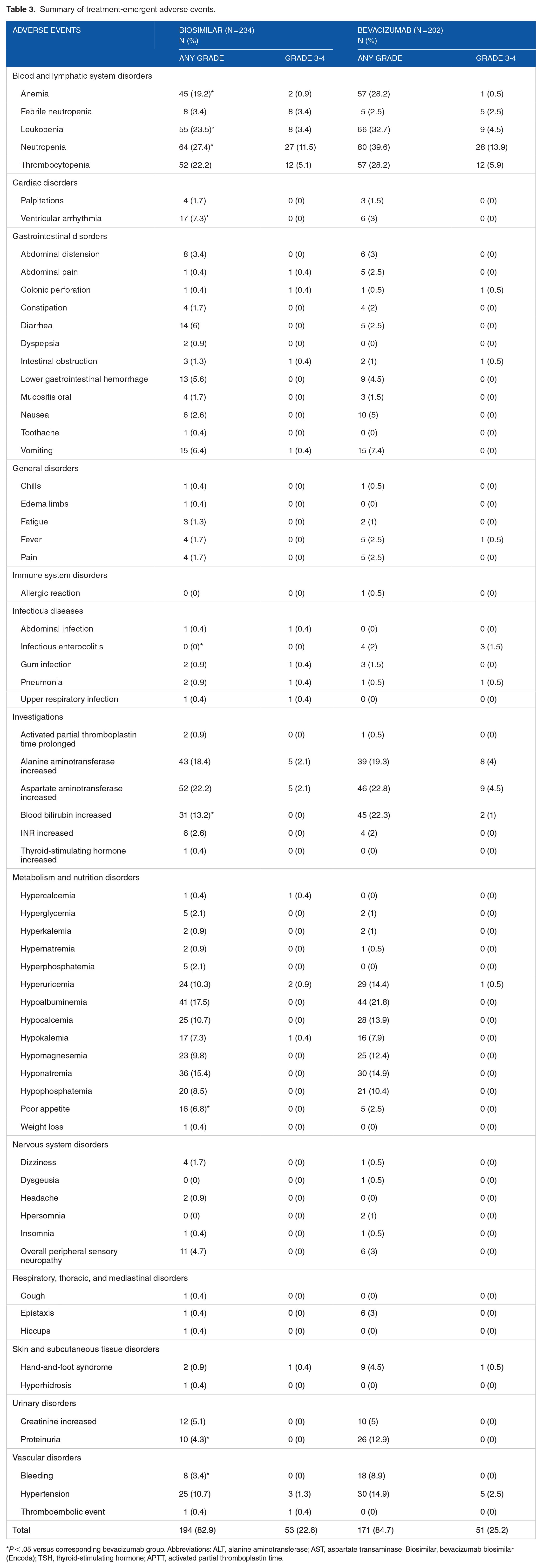
P < .05 versus corresponding bevacizumab group. Abbreviations: ALT, alanine aminotransferase; AST, aspartate transaminase; Biosimilar, bevacizumab biosimilar (Encoda); TSH, thyroid-stimulating hormone; APTT, activated partial thromboplastin time.
In the bevacizumab biosimilar (Encoda) group, neutropenia (27.4%) had the highest incidence of TEAEs, followed by leukopenia (23.5%), thrombocytopenia (22.2%), increase in aspartate transaminase (AST) (22.2%), anemia (19.2%), increase in alanine aminotransferase (ALT) (18.4%), hypoalbuminemia (17.5%), hyponatremia (15.4%), increase in blood bilirubin (13.2%), hypocalcemia (10.7%), hypertension (10.7%), and hyperuricemia (10.3%). In the bevacizumab group, neutropenia (39.6%) had the highest incidence of TEAEs, followed by leukopenia (32.7%), anemia (28.2%), thrombocytopenia (28.2%), increase in AST (22.8%), increase in blood bilirubin (22.3%), hypoalbuminemia (21.8%), increase in ALT (19.3%), hyponatremia (14.9%), hypertension (14.9%), hyperuricemia (14.4%), hypocalcemia (13.9%), proteinuria (12.9%), hypomagnesemia (12.4%), and hypophosphatemia (10.4%).
The incidence of anemia, leukopenia, neutropenia, ventricular arrhythmia, infectious enterocolitis, increase in blood bilirubin, poor appetite, proteinuria, and bleeding showed significant differences between the bevacizumab biosimilar (Encoda) group and bevacizumab group. The incidence rates of anemia (19.2% vs 28.2%), leukopenia (23.5% vs 32.7%), neutropenia (27.4% vs 39.6%), infectious enterocolitis (0% vs 2.0%), increase in blood bilirubin (13.2% vs 22.3%), proteinuria (4.3% vs 12.9%), and bleeding (3.4% vs 8.9%) at any grade in patients treated with bevacizumab biosimilar (Encoda) were significantly lower compared with those observed in patients treated with bevacizumab; however, no significant difference was found in the incidence rates of these AEs at grade 3/4. The incidence of ventricular arrhythmia (7.3% vs 3.0%) and poor appetite (6.8% vs 2.5%) at any grade in patients treated with bevacizumab biosimilar (Encoda) was significantly higher compared with those AEs in patients treated with bevacizumab. However, no difference was observed in the incidence of grade 3/4 ventricular arrhythmia and poor appetite.
Discussion
The present real-world study represents the first comparative analysis of the efficacy and safety profiles between bevacizumab biosimilar (Encoda) and bevacizumab as first-line therapy in patients with mCRC. Our findings demonstrate that bevacizumab biosimilar (Encoda) exhibits comparable efficacy to bevacizumab. The ORR risk ratio and risk difference fell within the limits stipulated by the NMPA, FDA, and EMA, signifying clinical equivalence between bevacizumab biosimilar (Encoda) and bevacizumab. The safety analysis revealed significant differences in the incidence of anemia, leukopenia, neutropenia, ventricular arrhythmia, infectious enterocolitis, increase in blood bilirubin, poor appetite, proteinuria, and bleeding between the bevacizumab biosimilar (Encoda) group and bevacizumab group. However, there were no statistically significant differences in grade 3 or 4 AEs between the bevacizumab biosimilar (Encoda) and bevacizumab groups.
Chemotherapy combined with targeted therapy constitutes the cornerstone of treatment for mCRC. 11 Bevacizumab, a monoclonal antibody that targets VEGF, combined with backbone chemotherapy effectively prolongs PFS and OS by inhibiting angiogenesis in patients with mCRC. This combination is currently considered the standard first-line treatment for RAS mutant patients with mCRC and those with tumors originating on the right side of the colon. 12
Biosimilars are highly similar to previously licensed biologics (also known as reference products or biologics) in quality, safety, and efficacy; however, they are not identical. 13 Biosimilars present an economically appealing alternative to costly reference biological drugs. 14 The first biosimilar of bevacizumab to enter the market was ABP215, which was approved by the FDA in 2018. 15 Encoda, a bevacizumab biosimilar, became the second one and was launched in China in 2019, making it the earliest bevacizumab biosimilar available in the Chinese market. 5 Bevacizumab biosimilar (Encoda) demonstrated a similar structure, in vitro tumor growth inhibition, and pharmacokinetic profile to bevacizumab.7,16 Approval of bevacizumab biosimilar (Encoda) for mCRC was based on the extrapolation principle applied to biosimilar. We aim to investigate the safety and efficacy of bevacizumab biosimilar (Encoda) in the treatment of mCRC through real-world experience, providing crucial practical insights for clinical oncologists and patients. In this study, the ORR risk ratio and risk difference fell within the prescribed limits set by regulatory authorities such as the NMPA, FDA, and EMA. This demonstrates that bevacizumab biosimilar (Encoda) is clinically equivalent to bevacizumab in the treatment of mCRC. Our study also revealed similar median PFS times between the 2 groups. Therefore, both bevacizumab biosimilar (Encoda) and bevacizumab exhibit comparable long-term efficacy in mCRC treatment. In clinical trials of other bevacizumab biosimilars as first-line treatment in patients with mCRC, the ORR ranges from 49% to 56%, while the median PFS is approximately 11 months. 17 Overall, our study showed a lower ORR but similar PFS duration compared with other clinical studies. The disparity in ORR may primarily stem from differences between real-world study and clinical trial.
The incidence rates of TEAEs at any grade (82.9% vs 84.7%) and those at grade 3/4 (22.6% vs 25.2%) in bevacizumab biosimilar (Encoda) group and bevacizumab group were similar without significant difference. No fatal effects were observed in either group. Neutropenia exhibited the highest incidence in both groups. The spectrum of very common AEs (>10%) was similar between the 2 groups. Most grade 3/4 TEAEs had low incidences in both treatment groups, except for neutropenia (>10%). In summary, both the AE spectra at any grade and AE incidence at grade 3/4 of bevacizumab biosimilar (Encoda) and bevacizumab were similar.
The incidence of anemia, leukopenia, neutropenia, ventricular arrhythmia, infectious enterocolitis, increase in blood bilirubin, poor appetite, proteinuria, and bleeding at any grade differed significantly between the bevacizumab biosimilar (Encoda) group and bevacizumab group. However, no significant difference in the incidence of grade 3/4 AEs was observed. In general, bevacizumab biosimilar (Encoda) and bevacizumab exhibit comparable safety profiles for grade 3/4 AEs, with only slight disparities in any grade AEs. We hypothesize that the limited sample size of this study may account for the observed safety variation. Future investigations involving a larger population are warranted to thoroughly evaluate potential safety discrepancies between these 2 drugs. However, we believe that the disparity in the incidence of grade 3/4 AEs holds greater clinical significance compared with any grade AEs. Consequently, this marginal discrepancy in safety profiles does not impact clinicians’ selection of therapeutic agents.
Admittedly, there are certain limitations associated with this study. First, the reliance on electronic health records for safety data may result in the absence of relevant information. Second, the sample size of this study is restricted. We eagerly anticipate a larger clinical study to further validate the clinical consistency of bevacizumab biosimilar (Encoda) and bevacizumab.
Conclusions
The present real-world study represents the pioneering evidence of comparable efficacy between bevacizumab biosimilar (Encoda) and bevacizumab in the first-line treatment of mCRC. Despite minor discrepancies in the safety data, the overall safety profile exhibited a similar pattern for both bevacizumab biosimilar (Encoda) and bevacizumab. Notably, bevacizumab biosimilar (Encoda) provides additional therapeutic options for patients with mCRC, particularly in economically disadvantaged regions.
Footnotes
Author Contributions
HS, JL, and QD conceived the idea. HS and SH led the primary data collection. HS, MW, HL, and SH analyzed the data. HS, MW, JL, and QD prepared the draft manuscript. HS, JL, and QD provided significant intellectual input in the redraft of the manuscript. All authors approved the final version of the manuscript.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by Shanghai Anticancer Association “Young eagle” program (Grant Number: SACA-CY21B04), Shanghai Anticancer Association “Soar” program (Grant Number: SACA-AX202110), and 2022 Shanghai Young Pharmaceutical Talent Ability Improvement Project.
Data Availability
All data analyzed in this research are available and can be provided.
Ethics Approval and Consent to Participate
This study was reviewed and approved by the Ethics Committee of Fudan University Shanghai Cancer Center and Zhongshan Hospital Fudan University (Approval Number: 2103232-10, B2021-049R) on April 14, 2021, and March 31, 2021. We have obtained written informed consent from all prospective subjects for publication of this study.