
Abstract
Metabolic reprogramming occurs when tumor cells replenish themselves with nutrients required for growth to meet their metabolic needs. Cancer-associated fibroblasts (CAFs) are activated fibroblasts involved in building the c (TME) to promote tumor progression and metastasis. Metabolic reprogramming of CAFs can interact with cancer cells to generate metabolic crosstalk. Furthermore, CAF metabolic reprogramming has great potential as a new field of tumor treatment. This review summarizes the role of CAFs in TME and the mechanisms by which metabolic reprogramming of CAFs causes cancer progression and metastasis, demonstrating the great potential of CAF metabolic reprogramming in cancer chemotherapy and immunotherapy treatment. Furthermore, we provide an outlook for future CAF metabolic reprogramming for cancer treatment.
Introduction
Tumor proliferation, invasion and metastasis are inseparable from the tumor microenvironment (TME). The TME is a complex ecosystem containing multiple cell types and noncellular components, including cancer cells, immune cells, endothelial cells, cancer-associated fibroblasts (CAFs), microvasculature, infiltrating biomolecules, and extracellular matrix (ECM). 1 These cells interact with cancer cells via complex communication networks (both autocrine and paracrine) in TME to induce tumor angiogenesis, immune evasion, apoptosis inhibition, drug resistance, and metabolic reprogramming. 2 Cancer cells can replenish nutrients required for growth by benefiting from metabolic adaptations obtained through various biomolecule synthesis, the provision of reducing equivalents, and adenosine triphosphate (ATP) production in the endocytic and extracellular pathways, tumor cells can meet the replenishment of nutrients required for growth, resulting in rapid and uncontrolled proliferation. Metabolic reprogramming refers to the process of adaptive changes in the metabolic properties of tumor cells through the conversion of available nutrients into biomass energy. 3
Tumor microenvironment is where metabolic reprogramming occurs most frequently, and signaling molecules, immune cells, fibroblasts, ECM, and peripheral vasculature can regulate tumor progression through metabolic reprogramming. 4 Activated fibroblasts, known as CAFs, constitute most stromal cells in the TME. 5 In contrast to normal fibroblasts (NFs), CAFs are involved in remodeling TME (such as cytokine, enzyme, and microRNA [miRNA] secretion, ECM track generation, and stromal stiffness enhancement) to promote tumor progression and metastasis. 5 Metabolic reprogramming of CAFs is defined as the reverse Warburg effect (RWE), characterized by an increase in lactate, glutamine, and pyruvate levels from aerobic glycolysis. 2 Metabolically reprogrammed CAFs replenish nutrients consumed by organelles (such as mitochondria) during tumor metabolism by generating energy at higher levels. In a recent study, Wu et al 6 discovered that transforming growth factor (TGF-β) RII can inhibit glucose metabolism in CAFs by attenuating Pyruvate kinase M2 (PKM2) nuclear translocation, thereby suppressing oral cancer tumor growth, indicating that targeting CAFs may be a new approach for cancer therapy. Furthermore, although the interactions between different cells in TME have received considerable attention, the complex relationship between the metabolic reprogramming of CAFs and cancer cells remains unknown and requires further investigation.
This review describes the current understanding of metabolic reprogramming of CAFs and their crosstalk with cancer cells. We emphasize potential therapeutic opportunities for targeting CAF metabolic reprogramming and focus on targeting CAFs strategies and immune cell metabolic crosstalk in cancer immunotherapy. In addition, we predicted the future of metabolic reprogramming of CAFs in cancer treatment.
Crosstalk Between CAFs and ECM: Promoting Immunosuppressive TME Formation
Extracellular matrix is a complex network composed of different macromolecules, including glycoproteins, fibronectin, proteoglycans, and collagen, and is responsible for maintaining normal tissue integrity, homeostasis, and growth. 7 Cancer-associated fibroblasts reshape ECM in several ways.8-10 First, CAFs can increase ECM matrix stiffness and degrade its normal structure by producing matrix metalloproteinases (MMP) (such as MMP-1 and MMP-3) and secreting matrix proteins (such as type I collagen and fibronectin), thus reshaping ECM. 8 Second, cytokine TGF-β1 released by CAFs enhances CAF invasiveness and stimulates ECM fibrosis by increasing smooth muscle α-actin (ACTA2), fibronectin, and laminin expression. 9 In addition, CAFs can upregulate various cytoskeletal regulators (such as diaphanous-related formin-3 [DIAPH3] and anillin (ANLN)) expressions through yes-associated protein (YAP) activation, inducing ECM stiffening. 11 Therefore, CAFs mediate mechanized fibrous barrier formation by fibrotic ECM, which is the basis for immunosuppressive TME formation. With intensive research, CAF-remodeled ECM was found to promote immunosuppressive TME formation by suppressing immune cell function and recruiting immunosuppressive cells, thus creating favorable conditions for tumor invasion and metastasis (Figure 1). 12
Cancer-associated fibroblast-remodeled ECM suppresses immune cell function. The ECM remodeled by CAFs acts as a physical barrier to inhibit the recruitment of immune cells to cancer sites by blocking immune cells (especially T cells) from cancer cells, thus impeding their participation in the immune response to TME and ultimately exerting an immunosuppressive function. 13 Cancer-associated fibroblast-remodeled ECM limits T-cell contact with cancer cells by increasing collagen deposition around tumor cell clusters, especially CD4+ T and CD8+ T cells. 14 This phenomenon, in which the infiltration of immune cells is inhibited and cannot kill tumors, is called tumor immune desertification, and how to reverse tumor immune desertification may be the key to improving the immunotherapy effectiveness. 15 Similarly, CAF-remodeled ECM can lead to poor infiltration of CD8+ cytotoxic T cells by exploiting the fibroregulatory effects of focal adhesion kinases (FAKs), thereby suppressing the immune response. 16 Furthermore, CAF-remodeled ECM leads to hypoxic TME formation by accumulating matrix proteins, inducing the soluble factor vascular endothelial-derived growth factor (VEGF) production to reduce the accumulation rate of circulating T-cells through the tumor vasculature, ultimately reducing their infiltration. 17 Therefore, CAF-remodeled ECM can promote immunosuppressive microenvironment formation by suppressing immune cell functions. However, recent studies have demonstrated that CAF-remodeled ECM suppresses T-cell function. Further studies are needed to determine the suppressive effect of CAF-remodeled ECM on other immune system cell functions.
Cancer-associated fibroblast-remodeled ECM recruits immunosuppressive cells. In the TME, immunosuppressive cells indirectly block immunotherapy through secreted factors that establish immune tolerance. 18 Cancer-associated fibroblast-remodeled ECM with increased collagen density recruits TAMs, myeloid-derived suppressor cells (MDSCs), and regulatory T cells (T-regs), leading to direct exhaustion of CD8+ T cells by activating intracellular FAKs, thereby triggering immunosuppression. 19 Cancer-associated fibroblast-remodeled ECM promotes M2-type polarization of macrophages and recruitment of tumor-associated macrophages (TAMs) by inducing oncogenic collagen matrices, thereby promoting immunosuppressive microenvironment formation. 20 Furthermore, fibrotic ECM alters factors affecting macrophage polarization in TME with the assistance of hyaluronic acid in a tumor model of CAK-I kidney cancer cells, thus favoring the enhancement of M2 phenotype and pro-tumor properties in macrophages, laying the foundation for TAM to enhance cell migration, invasion, and angiogenesis in the TME. 21 Therefore, CAFs-remodeled ECM can promote immunosuppressive TME formation by recruiting immunosuppressive cells. However, current studies on the recruitment of immunosuppressive cells by CAF-remodeled ECM are limited. Future research may determine whether CAFs-remodeled ECM can recruit immunosuppressive cells through signaling pathways and cytokines.

CAFs-remodeled ECM promotes the formation of an immunosuppressive microenvironment in 2 ways. (1) CAFs-remodeled ECM suppresses the function of immune cells. (2) CAFs-remodeled ECM recruits immunosuppressive cells. The figure was created by Figdraw (www.figdraw.com).
Metabolic Crosstalk Between CAFs and Cancer Cells
Cancer-associated fibroblasts in TME promote an immunosuppressive microenvironment conducive to cancer cell growth and generate metabolic crosstalk with cancer cells. Cancer cells reprogram CAF metabolism by absorbing secreted metabolites such as lactate and pyruvate. 22 Metabolic reprogramming of CAFs promotes malignant tumor progression by secreting ECM proteins and providing energy support. 22 Therefore, understanding the specific mechanisms of metabolic crosstalk between CAFs and cancer cells may be a good way to solve the dilemma currently encountered in cancer therapy.
Cancer cells mediate the metabolic reprogramming of CAFs
Cancer cells cannot grow without sufficient protein, lipid, nucleic acid, and ATP, which must be obtained through metabolic reprogramming. 23 Cancer cells metabolic reprogramming is a key way to maintain a hypo-differentiated state and cell proliferation, but it cannot completely satisfy growth needs, requiring the manipulation of CAFs to form a compensatory environment that provides metabolic intermediates for nutrient synthesis. 24 Cancer cells can mediate CAF metabolic reprogramming through 3 metabolic types: glucose metabolism, amino acid metabolism, and lipid metabolism, thereby promoting their growth, metastasis, and immune escape. 25
Metabolic reprogramming of glucose in CAFs
The Warburg effect refers to glycolysis in cancer cells under aerobic conditions, resulting in increased glucose uptake and secretion of lactate and pyruvate. 26 Like the Warburg effect, CAFs exhibit changes in glucose metabolism and aerobic glycolysis that are mediated by cancer cells, referred to as the reverse “Warburg effect..” 26 The reverse “Warburg effect” suggests that CAFs can provide nutrients to mitochondrial oxidative phosphorylation (OXPHOS) in neighboring cancer cells through aerobic glycolysis. Currently, 2 potential mechanisms are associated with the metabolic reprogramming of glucose in CAFs, resulting in a CAFs glycolytic phenotype (Figure 2). First, CAFs enhance glycolysis by contacting the cancer cells. Cancer cells activate the p27 protein and G1/S checkpoint expression, and inhibit cyclin-dependent kinase 2 (CDK2) by upregulating 6-phosphofructokinase liver type (PFKL) and hexokinase 2 (HK2) in CAFs, thus linking the cell cycle to glycolysis and ultimately reprogramming glucose. 27 The oncogene c-Myc upregulates glucose transporter protein 1 (GLUT-1) and lactate dehydrogenase-A (LDH-A) expression by transactivating the LDH-A promoter, thereby enhancing glucose uptake and metabolic flux in CAFs. 28 In addition, small extracellular vesicles (sEVs) secreted by cancer cells increase GLUT-1 levels in CAFs by downregulating miR-186 levels, thereby enhancing glycolysis. 29 Small extracellular vesicles are secreted by almost all cells, surrounded by a lipid bilayer, and carry various biomolecules, including DNA, RNA, lipids, glycans, proteins, and metabolites, providing a delivery vehicle for metabolic crosstalk between cancer cells and CAFs. 30 Second, CAFs produce lactate and pyruvate when in contact with cancer cells. In osteosarcoma cells, cancer cells use monocarboxylate transporter 1 (MCT-1) to deliver lactate to CAFs for metabolic energy. 31 Cancer cells in the bladder and breast produce excess lactate and upregulate MCT-4 expression in CAFs by activating MMP-9, thus enhancing the CAF ability to take up glucose. 32 Moreover, pancreatic cancer cells increase lactate dehydrogenase, pyruvate kinase, and mRNA expression in CAFs by upregulating succinate dehydrogenase (SDH), fumarate hydratase (FH), and MCT1 levels, increasing lactate production and glucose uptake in CAFs and ultimately promoting pancreatic cancer invasion and migration. 33 Notably, Cavilin-1 (Cav-1) is a membrane-bound scaffold protein that partitions and negatively regulates signal transduction. 34 Proteomic analysis revealed that loss of Cav-1 expression in CAFs upregulates LDH-B and PKM2 levels, activating the TME and providing lactate and pyruvate for cancer cell growth, ultimately promoting the development and metastasis of prostate and breast cancers. 35 This may be related to the fact that Cav-1 deficiency promotes paracrine signaling mechanisms, including fibrosis, ECM remodeling, and CAF metabolic reprogramming. However, the mechanism by which cancer cells and Cav-1-deficient CAFs produce metabolic crosstalk remains to be investigated. Taken together, cancer cells can mediate metabolic reprogramming of glucose in CAFs to provide energy for their progression and metastasis.
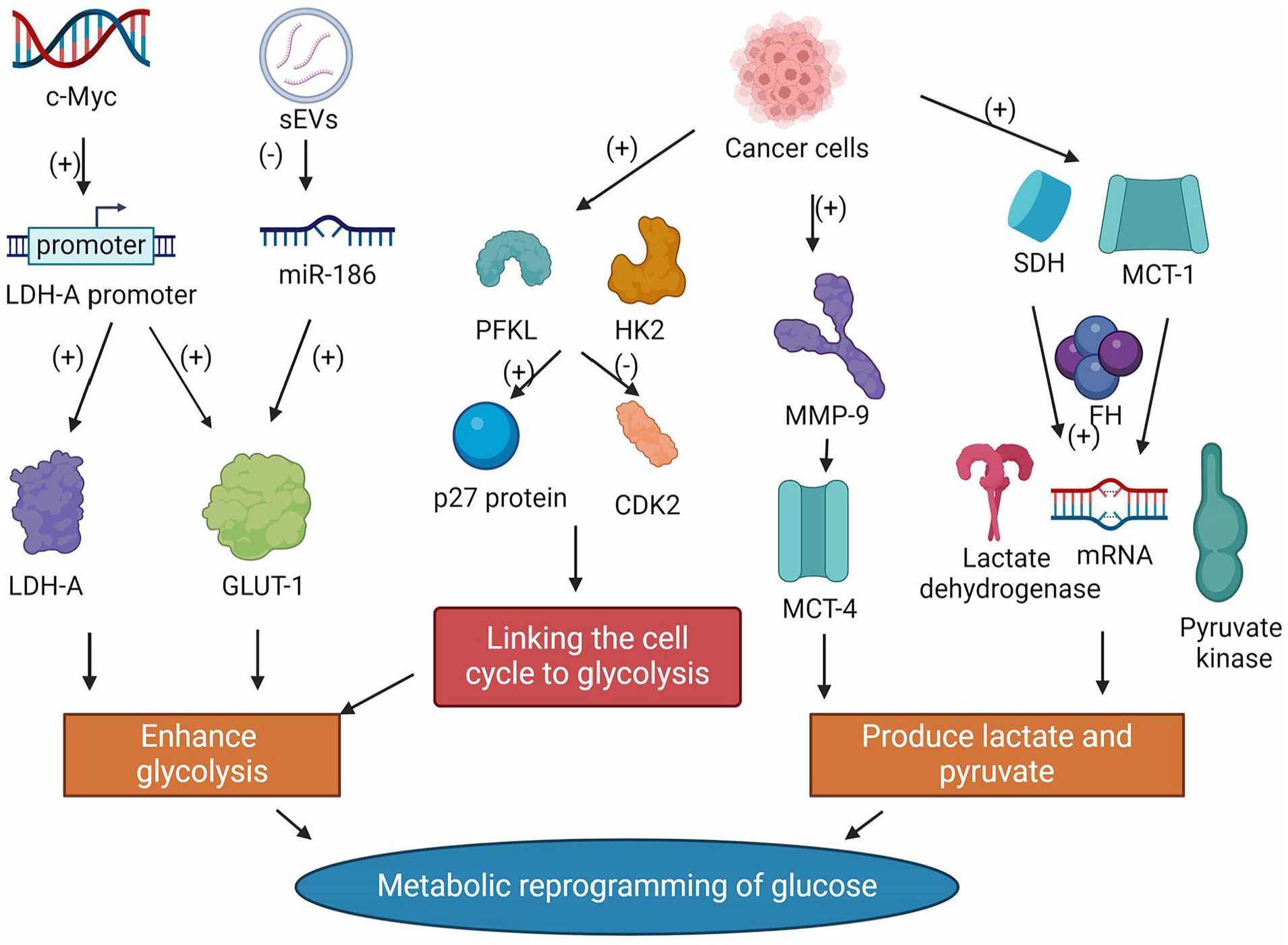
Metabolic reprogramming of glucose in CAFs. Metabolic reprogramming of glucose in CAFs. Created with BioRender.com.
Metabolic reprogramming of amino acids in CAFs
Cancer-associated fibroblasts can enhance the output of amino acids as an energy source, in addition to glucose, to meet the demands of the rapid proliferation of cancer cells (Figure 3). Glutamine (Gln), the most widely studied amino acid, is a major carbon and nitrogen source that is essential for mitochondrial metabolism in cancer cells. 36 Glutamine fuels tumor cell mitochondria by supplementing the tricarboxylic acid (TCA) cycle with oxaloacetic acid and accumulating intermediates, thereby promoting rapid cancer cell proliferation, which makes Gln a more critical source of ATP than glucose. 37 Moreover, Gln is essential for maintaining cancer cell integrity and mitochondrial membrane potential. 38 Cancer-associated fibroblasts, mediated by cancer cells, are required for cancer cell invasion and metastasis through upregulation of Gln metabolism. Sazeides et al 39 found significantly higher levels of 13C-labeled m+ 5α-KG and m+ 5 glutamate in 13C5-glutamine labeling experiments while studying the relationship between sEVs and mitochondria in prostate cancer patients, indicating that prostate cancer cells can mediate CAFs to upregulate Gln metabolism and alter their metabolic patterns, thus satisfying the dependence of sEVs and mitochondria on Gln. In ovarian cancer, cancer cells mediate CAFs to produce large amounts of Gln using glutamine synthetase (GS), which sets the stage for further conversion of Gln to glutamate and into the TCA cycle, ultimately promoting tumor progression. However, more studies are needed to demonstrate whether glutamine can promote CAF metabolic reprogramming in other cancers, and the mechanisms involved remain unclear. 40 In addition to Gln, CAFs produce other amino acids as nitrogen sources that provide nutrition to cancer cells. In p62k CAFs, despite the lack of Gln, its expression through upregulation of pyruvate carboxylase (PC) and asparagine synthetase (ASNS) can regulate pyruvate and aspartate synthesis, thereby enhancing the TCA cycle and sustaining cancer cell survival. 41 In addition, human skin squamous carcinoma (HSSC) cells mediate glutamate and aspartic acid utilization by CAFs through the solute carrier family 1 membrane 3 (SLC1A3) channel, thus promoting cancer cell proliferation and biosynthesis. 42 In summary, CAFs provide important energy support for malignant cancer progression by altering their amino acid metabolic levels.

Three metabolic types associated with the metabolic reprogramming of CAFs. Metabolic reprogramming of amino acids in CAFs. Metabolic reprogramming of lipid in CAFs. Created with BioRender.com.
Metabolic reprogramming of lipid in CAFs
Lipids are natural compounds that are insoluble in water but soluble in nonpolar solvents. 43 Fatty acids (FAs) are important lipids, because they serve as structural components of cell membranes and second messengers involved in intracellular signal transmission. 44 Additionally, it is a source of energy when scarce or restricted. 44 Several recent studies have demonstrated that dysregulated lipid metabolism in CAFs, mediated by cancer cells, can promote tumor growth and metastasis.45-47 In ovarian cancer, lysophosphatidic acid (LPA) derived from cancer cells mediates intracellular liposome remodeling in CAFs through hypoxia-inducible factor (HIF-1α) upregulation by the LPA receptor (LPAR), thereby promoting tumor proliferation and metabolism. 45 In colorectal cancer (CRC), CAFs increase FA production by activating fatty acid synthase (FASN), leading to lipid metabolism reprogramming and promoting uptake by CRC cells, which enhances the metastatic ability of CRC cells. 46 Moreover, in breast cancer, CAFs promote the uptake of exogenous fatty acids by cancer cells by upregulating fatty acid transporter protein 1 (FATP1) upregulation in human MDA-MB-231 TNBC cells. 47 Thus, cancer cells can mediate lipid metabolic reprogramming in CAFs to provide energy for progression and metastasis. However, recent studies have demonstrated that the mechanisms promoting lipid metabolism reprogramming in CAFs vary in different cancers. Future research should investigate whether mechanisms applicable to most cancers can mediate lipid metabolism reprogramming.
CAF metabolic reprogramming affects tumor progression
Cancer-associated fibroblast metabolic reprogramming promotes tumor progression by reshaping the metabolic levels of cancer cells in 3 important ways: exporting nutrients, regulating metabolic enzymes, and suppressing immune responses (Table 1).
Metabolic reprogramming of CAFs promotes tumor progression in multiple ways.

Abbreviations: CAF, cancer-associated fibroblast; CREB, cAMP response element-binding protein; FAK, focal adhesion kinase; HGF, hepatocyte growth factor; HNSCC, head and neck squamous cell carcinomas; IDO, indoleamine 2,3-dioxygenase; MDSC, myeloid-derived suppressor cell; PGM, phosphoglucomutase; PKM, pyruvate kinase M; sEVs, small extracellular vesicles; STAT3, signal transducer and activator of transcription 3; TGF, transforming growth factor; WNK1, WNK Lysine Deficient Protein Kinase 1.
CAFs promote tumor progression by exporting nutrients
During metabolic reprogramming, CAFs produce numerous nutrients that cancer cells absorb to promote tumor growth. Small extracellular vesicles, approximately 100 nm in diameter, contain biologically active components such as proteins, lipids, miRNAs, and mRNAs, which change the biological properties of the recipient cells and mediate the exchange of substances between cells. 48 Recent studies have discovered that sEVs act as delivery vehicles in the metabolic crosstalk between CAFs and cancer cells, where they are internalized by cancer cells and transfer metabolites (such as proteins, mRNAs, miRNAs, and long non-coding RNA [lncRNAs]) from CAFs to tumor cells, thereby promoting tumor progression and metastasis.49-51 In 13C isotope labeling experiments, prostate cancer CAF-derived sEVs increased glutamate-dependent glycolysis and reductive carboxylation in cancer cells by inhibiting OXPHOS in mitochondria, thereby providing amino acids to nutrient-deficient cancer cells to promote tumor progression. 49 Similarly, ultrahigh-performance liquid chromatography (UPLC) and gas chromatograph-mass spectrometry (GC-MS) experiments demonstrated that prostate cancer CAF-derived sEVs are rich in metabolites, such as TCA cycle intermediates, amino acids, and lipids, which are used as nutrients for tumor progression. 50 In addition, lymphoma CAF-derived sEVs can increase glycolysis and ATP levels, thereby promoting lymphoma cell growth. 51 However, the metabolites carried by lymphoma CAF-derived sEVs and their relationship with glycolysis have not been investigated; this may be demonstrated in the future. In addition to direct nutrient export, CAF-derived sEVs can promote glutamate-dependent reductive carboxylation and glycolysis in cancer cells by transferring substrates and noncoding RNAs (ncRNAs). Noncoding RNAs, including the previously mentioned circular RNA (circRNA), lncRNA, miRNA, and PIWI-interacting RNA (piRNA), can act as suppressors or oncogenes to influence malignancy onset, progression, and metastasis. Breast cancer CAF-derived sEVs can inhibit mitochondrial OXPHOS by increasing PKM expression and decreasing miR-330-5p expression, thereby increasing glycolysis and reductive carboxylation, ultimately enhancing breast tumor cell proliferation. 55 This indicates that ncRNAs may be targets for regulating metabolic crosstalk between cancer cells and CAFs, and in-depth studies may provide new directions for cancer therapy.
CAFs promote tumor progression by regulating metabolic enzymes
Cancer cells mediate CAF metabolic reprogramming and induce CAFs to regulate key metabolic pathway enzymes by secreting various cytokines, thereby promoting tumor proliferation and metastasis. Cancer cell-mediated CAFs use TGF-β to promote glycogenolysis and induce the phosphorylation of phosphoglucomutase 1 (PGM1) by secreting CXCL10, interleukin (IL)-6, and IL-8, thereby activating the pentose phosphate pathway (PPP) and promoting glycolysis in ovarian cancer cells, enhancing the cancer cell ability to invade and metastasize. 52 Similarly, CAFs upregulate the glycolytic enzyme LINC00092 through high CXCL14 expression in vitro and in vivo, thereby enhancing glycolysis and metabolism in ovarian cancer cells, ultimately promoting tumor metastasis. 53 In head and neck squamous cell carcinomas (HNSCC), CAFs affected by lactate upregulates glycolytic enzymes (phosphofructokinase and hexokinase II) expression by secreting hepatocyte growth factor (HGF), thereby promoting glycolysis in cancer cells and tumor progression. 56 Moreover, CAFs increase Ccl6 and Ccl12 secretion and activate protein kinase A (PKA) in malignant cells by depleting FAK, thus enhancing paracrine signaling of tumor proliferation and cancer cell glycolysis. 54 A recent study found that CAFs regulate metabolism-related signaling pathways in cancer cells. Insulin-like growth factor 1 (IGF-1) secreted by CAFs can activate the mammalian target of rapamycin (mTOR) pathway by binding to the IGF-1 receptor on CRC cells, thus triggering lactate release and glucose uptake, which promotes Gln uptake and cellular channel protein SLC7A11 (solute carrier family 7, membrane 11) expression in CRC cells. This indicates that altering signaling pathways may promote tumor progression by affecting cancer cell metabolism, such as altering metabolic enzymes; however, additional relevant mechanisms remain to be discovered.
CAFs promote tumor progression by suppressing immune responses
Reprogrammed CAFs promote tumor progression by suppressing the immune response in 3 major ways. First, metabolic reprogramming of CAFs produces metabolites that suppress the immune response. Cancer-associated fibroblasts inhibit CD8+ T-cell toxicity and induce T-cell anergy by secreting indoleamine 2,3-dioxygenase (IDO). 57 In lung cancer, CAFs complex WNK Lysine Deficient Protein Kinase 1 (WNK1) and cAMP response element-binding protein (CREB) by secreting kynurenine and overexpressing tryptophan 2,3-dioxygenase (TDO2), thus inhibiting differentiated dendritic cells, ultimately leading to immunosuppression. 58 In addition, arginase II (ARG2) secreted by CAFs can suppress the immune response by blocking antitumor T-cells. 59 Second, reprogrammed CAFs can suppress immune responses by interacting with neutrophils. Cancer-associated fibroblasts that undergo cancer cell-mediated reprogramming suppress neutrophils via the IL6/signal transducer and activator of the transcription 3 (STAT3) axis and CCL2-CCR-2 axis, thereby enhancing mesenchymal stem cell (MSC) transformation and tumor proliferation. 60 In hepatocellular carcinoma, CAFs can lead to apoptosis of neutrophils through the activation of c-Jun kinase and STAT3-programmed cell death ligand 1, promoting tumor growth. 61 Finally, metabolically reprogrammed CAFs induce hypoxic environment formation by participating in aberrant angiogenesis, thereby promoting immunosuppression of TME. Cancer-associated fibroblasts impede T-cell infiltration by secreting pro-angiogenic factors, suppressing T-cell function, and causing immunosuppression. 62 In addition, CAFs in hypoxic environments enhance programmed death ligand 1 (PD-L1) expression in MDSCs by binding to the PD-L1 proximal promoter and HIF-1α, causing cancer cells to evade immune surveillance. 63
Metabolic Reprogramming of CAFs Is Involved in Cancer Therapy
Chemotherapy strategies targeting the metabolic reprogramming of CAFs
Despite the significant clearance of chemosensitive tumors by chemotherapy, many tumor cells remain viable owing to tumor heterogeneity (Figure 4). Tumor heterogeneity refers to the specific phenotypic characteristics of different cell populations in tumors, including intratumor, intermetastatic, intrametastatic, and interpatient tumor heterogeneity. This is the primary reason for the survival of tumor cells with intrinsic or acquired drug resistance. 64 Surviving tumor cells induce CAFs to secrete chemokines by mediating the metabolic reprogramming of CAFs, thereby enhancing invasiveness and resistance to chemotherapeutic agents, ultimately leading to tumor drug resistance. 65 Currently, several potential chemotherapeutic strategies targeting the metabolic reprogramming of CAFs may improve the dilemma encountered in cancer chemotherapy. Targeting metabolically reprogrammed CAFs using antibodies against cell surface markers (GPR77 and CD10) inhibits NF-κB activation and blocks GPR77 complement signaling caused by p65 acetylation and phosphorylation, thereby improving chemosensitivity in lung and breast cancers. 66 Docetaxel, in combination with smoothened inhibitors, can inhibit the activation of Hedgehog signaling by CAFs, thereby impeding CAF metabolic reprogramming and tumor cell resistance phenotype expression. 67 Similarly, gemcitabine inhibits fibrosis and stromal inflammation caused by CAF metabolic reprogramming by combining with vitamin D receptor (VDR) ligands, improving the pancreatic tumor ability to uptake gemcitabine and enhancing survival in pancreatic cancer patients. 68 Moreover, chemotherapy combined with a DNA vaccine targeting fibroblast activation proteins (FAPs) enhances CD8T cells ability to kill CAFs, thereby improving the body’s ability to absorb chemotherapeutic drugs. 69 Interestingly, a recent study discovered that rhythmic therapy, in which low doses are administered on a frequent or continuous schedule, can inhibit CAFs from secreting paracrine signals for metabolic reprogramming, thereby inhibiting tumor progression and enhancing chemotherapeutic efficacy. 70 However, these strategies are still experimental and have not yet been tested for their clinical relevance. Further research is needed to determine whether these strategies apply to clinical treatment.
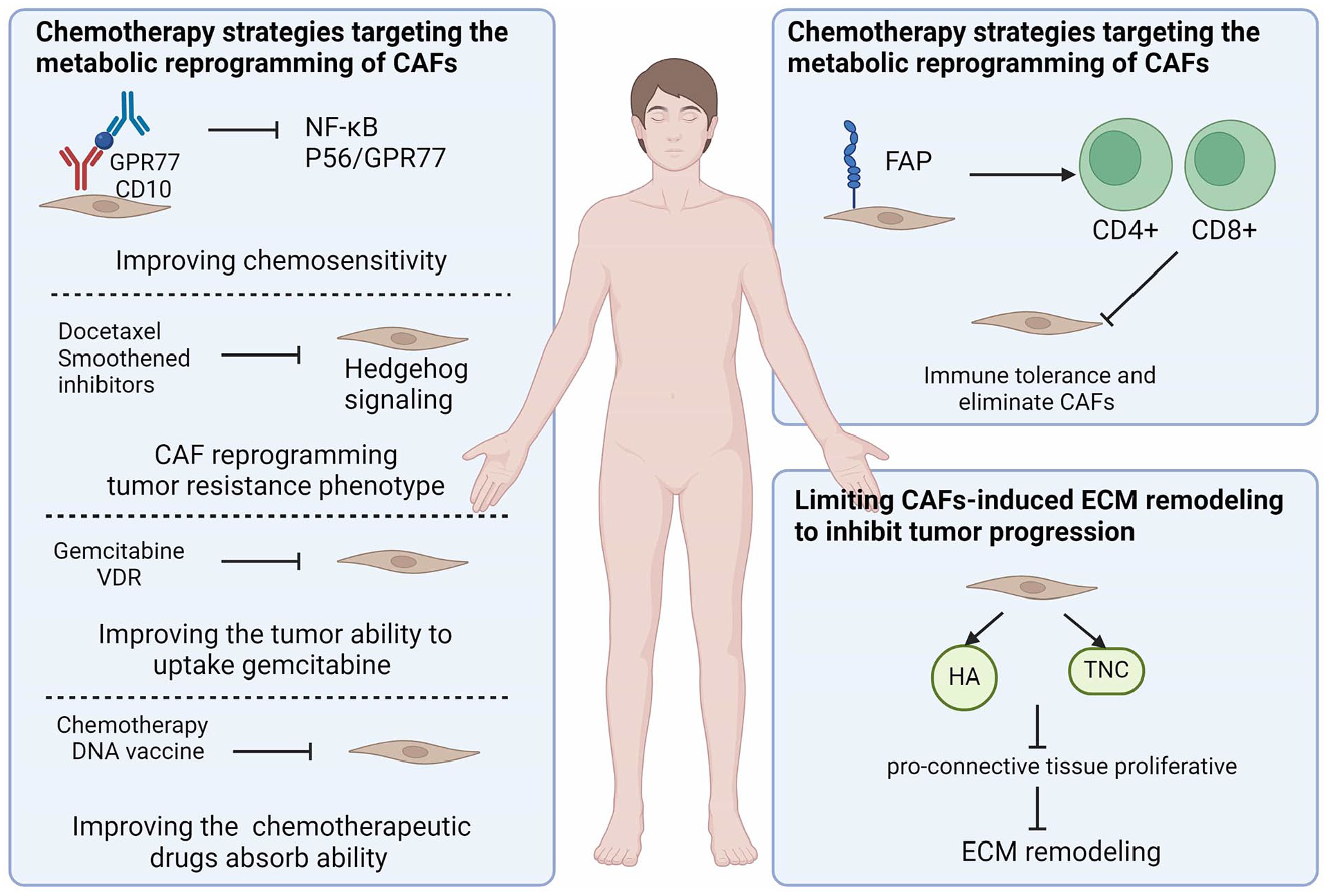
Metabolic reprogramming of CAFs is involved in cancer therapy. (1) Chemotherapy strategies targeting the metabolic reprogramming of CAFs; (2) immunotherapeutic strategies targeting the CAF metabolic reprogramming: (1) Direct action on CAFs targets to deplete CAFs; (2) Limiting CAFs-induced ECM remodeling to inhibit tumor progression. Created with BioRender.com.
Immunotherapeutic strategies targeting the CAF metabolic reprogramming
Immunotherapy is emerging as one of the most promising methods for targeting the metabolic reprogramming of CAFs to inhibit tumor proliferation by investigating the link between CAF metabolic reprogramming and the immunosuppressive microenvironment. 71 There are 2 potential immunotherapeutic strategies to inhibit tumor progression by targeting CAF metabolic reprogramming: direct action on CAFs targets to deplete CAFs and restriction of CAF-induced ECM remodeling.
Direct action on CAFs targets to deplete CAFs
Immunotherapeutic strategies that act directly on CAFs deplete CAF metabolic reprogramming using inhibitors of CAF surface markers. Fibroblast activation protein, the most popular surface marker for CAFs, enhances the toxic effects of CD8+ T-cells and promotes tumor necrosis factor (TNF)-α and interferon (IFN)-γ-mediated immune responses through targeted therapies, including tumor vaccines and T-cell therapy, thereby eliminating FAP+ cells and CAFs. 72 First, FAP-based DNA vaccines disrupt immune tolerance and eliminate CAFs by promoting CD4+ and CD8+ T-cell-mediated immune responses, thereby inhibiting tumor progression, 73 . 74 Second, FAP-based DC vaccines impede Treg cells migration to tumors by reducing TGF-β levels and enhancing T-cell responses, thereby inhibiting CAFs with high FAP expression and ultimately suppressing tumor growth and metastasis.75,76 Furthermore, FAP-based T-cell therapy depletes FAP-positive cells using chimeric antigen receptor (CAR) T-cells, thereby impeding tumor stroma formation and facilitating the uptake of antitumor chemotherapeutic agents (such as gemcitabine). 77 However, considerable challenges remain concerning FAP-based therapies. Certain tumor vaccines, such as peptide immune vaccines, whole-cell vaccines, and adenoviral vector vaccines, are not the same for CAFs, even though they can destroy FAP+ stromal cells. The mechanisms by which other FAP-targeted therapies, including FAP-targeted liposomes, immunotoxins, antibodies, and cisplatin, affect metabolic reprogramming of CAFs remain to be investigated. Notably, α-SMA, another surface marker of CAFs, is closely associated with CAF metabolic reprogramming. 78 In pancreatic and breast cancers, Özdemir et al 78 discovered that attenuated infiltration of CD3+ Foxp3+ Treg cells enhanced myofibroblast depletion and inhibited fibrosis, thereby depleting α-SMA + CAFs, which inhibited tumor progression and angiogenesis. However, the accuracy of FAP- and α-SMA-based immunotherapy is unsatisfactory, and highly selective surface markers of CAFs need to be discovered and investigated in depth. Table 2 displays the clinical trials of CAF metabolic reprogramming by FAP-targeted therapy.
Clinical trials related to metabolic reprogramming of CAFs by FAP-targeted therapy.

Limiting CAFs-induced ECM remodeling to inhibit tumor progression
Considering that CAFs can promote the formation of an immunosuppressive microenvironment by remodeling the ECM, thus favoring tumor invasion and metastasis, inhibiting tumor progression by limiting CAF-induced ECM remodeling is a feasible strategy for immunotherapy. Hyaluronan (HA) and Tenascin C (TNC), the predominant CAF-derived ECM proteins, have been demonstrated to be important targets for inhibiting ECM remodeling by attenuating the pro-connective tissue proliferative response. 79 Human recombinant PH20 hyaluronidase (PEGPH20) depletion of HA by combining nab-paclitaxel and gemcitabine can reduce vascular compression and improve vascular patency, thereby inhibiting ECM remodeling, which promotes immune cell entry into the tumor vasculature and inhibits tumor progression. 80 Losartan inhibits the HA production and stromal collagen by inhibiting the compliance signaling of endothelin-1 (ET-1), connective tissue growth factor (CTGF), and TGF-β1, thereby reducing remodeled ECM component formation and decreasing the immunosuppressive effect of ECM on immune cells. 81 Furthermore, antibody F16 recruits immune cells to the TME. It downregulates TNC expression in cancer tissues by complexing IL-2, thus reducing the signaling of effector molecules to TNC-rich tumor tissues, and ultimately inhibiting tumor proliferation and invasion. 82
Future Prospects of Targeting CAF Metabolic Reprogramming to Treat Cancer
Numerous findings have linked CAF metabolic reprogramming to cancer development, progression, and metastasis. As a new field, CAF metabolic reprogramming has great potential for tumor treatment. However, current studies on CAF metabolic reprogramming are limited to tumor model studies, leading to limited data regarding its use in clinical treatment. In the future, metabolic reprogramming targeting CAFs appears to have great potential to play a role in the clinical treatment of cancer. They may focus on the following promising aspects.
Conduct preclinical studies to determine the mechanism underlying the combined effects of multiple metabolic inhibitors and immunotherapy in CAFs. By further understanding this combined mechanism helps to improve the adjuvance of CAFs for immunotherapy, both from the perspective of reducing tumor promotion and affecting immune cells.
In-depth exploration of the impact of metabolic reprogramming of glucose, amino acids and lipids in regulating CAFs in cancer. A detailed understanding of the mechanisms can help in the development of drugs targeting relevant CAFs and their utilization.
The relationship between immune cells and CAF metabolic reprogramming remains to be understood. Immune cells are key to influencing TME, and exploring the relationship between immune cells and CAFs can further clarify the relevant mechanisms between them and tumorigenesis and development.
The role of CAF metabolic reprogramming in radiotherapy has not yet been discovered, and whether there is an association between them is also a question worth exploring. Given the relationship of CAF to chemotherapy and immunotherapy, and its associated role in TME, we hypothesized that CAF would also be able to influence chemotherapy for tumors.
While we describe the mechanisms by which metabolic reprogramming leads to tumor progression and metastasis, and its potential in chemotherapy and immunotherapy. However, this article still has certain shortcomings. In particular, it lacks the study of related clinical trials, because the current off related studies still remain at the preclinical level and need to be further explored and researched.
Conclusion
Given that CAFs have complex crosstalk with numerous cellular structures within the TME, metabolic reprogramming of CAFs plays an important role in cancer cell growth, metastasis, and immune reprogramming. Therefore, therapies targeting reprogramming of CAFs are important in oncology, especially in combination with chemotherapy and immunotherapy. However, despite the success of these inhibitors in tumor models, breakthroughs in clinical translation still require more in-depth research in related directions.
Footnotes
Acknowledgements
The Figure was created by Figdraw (www.figdraw.com) and BioRender.com. The authors thank Home for Researchers editorial team (![]() ) for language editing service.
) for language editing service.
Author Contributions
LZ and WZ drafted the manuscript in detail. WZ and LZ researched the literatures and drawed figures. XH and LZ counted and plotted the diagram and table. DT and DW critically revised the article for important intellectual content. All authors read and approved the final manuscript.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Graduate Research-Innovation Project in Jiangsu province (grant no. SJCX21_1644), the Academic Science and Technology Innovation Fund for College Students (grant no. 202011117056Y), the Social Development-Health Care Project of Yangzhou, Jiangsu Province (grant no. YZ2021075), and High-level talent “six one projects” top talent scientific research project of Jiangsu Province (grant no. LGY2019034), the Graduate Research-Innovation Project in Jiangsu province (grant no. SJCX22_1816), and Social development project of key R & D plan of Jiangsu Provincial Department of science and technology (grant no. BE2022773). The funding bodies had no role in the design of the study; in the collection, analysis, and interpretation of the data; and in the writing of the manuscript.
Availability of Data and Material
Not applicable
Ethics Approval and Consent to Participate
Not applicable
Consent for Publication
Not applicable