
Abstract
Complex glycerol kinase deficiency (CGKD), also known as Xp21 contiguous gene deletion syndrome, is a rare X-linked recessive disorder resulting from partial deletion of the Xp21.3 chromosomal region. CGKD encompasses several loci, including glycerol kinase (GK), Duchenne muscular dystrophy (DMD), X-linked adrenal hypoplasia congenita (NR0B1), and intellectual developmental disorder (IL1RAPL1). We present the cases of two male siblings diagnosed with CGKD. The elder sibling was initially suspected of having congenital adrenal hypoplasia (CAH). Whole exome sequencing (WES) revealed an interstitial deletion of 6.6 Mb on Xp21.3p21.1, encompassing critical genes including GK, DMD, NR0B1, and IL1RAPL1. The younger sibling was diagnosed shortly after birth based on family history, clinical and biochemical findings. The presented report highlights the diagnostic challenges associated with CGKD and the important role of genetic testing in confirming the diagnosis. A multidisciplinary team approach is necessary.
Plain Language Summary
Complex Glycerol Kinase Deficiency (CGKD) is a rare, inherited condition caused by the deletion of a group of genes on the X chromosome, affecting various systems in the body, such as muscles, hormone regulation, and development. Diagnosing CGKD is challenging due to its diverse and complex symptoms, as illustrated in this case of two brothers. The older sibling showed early signs of adrenal insufficiency, including poor feeding, vomiting, and abnormal skin pigmentation, and was initially misdiagnosed with a more common adrenal condition. Genetic testing later confirmed CGKD through the identification of a specific X chromosome deletion. This delayed diagnosis emphasizes the need to consider rare genetic disorders when symptoms are atypical. The younger brother was diagnosed at birth through family history, clinical evaluations, and genetic testing, enabling early management with hormone replacement therapy and dietary adjustments. Both children are now supported by a multidisciplinary team, addressing their medical, developmental, and nutritional needs, which has improved their health and quality of life. This case highlights the importance of genetic testing in diagnosing CGKD, especially in cases of unexplained developmental delay and adrenal insufficiency, as early detection allows for better management and family support.
Keywords
Introduction
Glycerol kinase deficiency (GKD, OMIM: 307030) is a rare X-linked recessive disorder caused by pathogenic variants in GK on chromosome Xp21. Three forms of GKD are recognized: infantile, juvenile, and adult forms. While the juvenile and adult forms are regarded as isolated GKD, the infantile form manifests itself as part of the complex glycerol kinase deficiency (CGKD). CGKD (also called chromosome Xp21 deletion syndrome, OMIM: 300679) belongs to a group of contiguous gene syndromes (CGS), caused by partial deletion of the Xp2.1 region. Currently, more than 100 cases of CGKD are described. 1 Usually, CGKD encompasses genes associated with GKD (GK), Duchenne muscular dystrophy (DMD), X-linked adrenal hypoplasia congenita, AHC (NR0B1), and intellectual developmental disorder, X-linked 21 (IL1RAPL1). The AHC and DMD loci are the closest to the glycerol kinase deficiency (GKD) locus, making the combined AHC-GKD-DMD the most common genotype. 2 However, the loci for chronic granulomatous disease, retinitis pigmentosa, Aland Island eye disease, McLeod phenotype, ornithine transcarbamylase deficiency, CFAP47 (cilia- and flagella-associated protein 47), CYBB (gp91), XK (Kell antigen), RPGR (retinitis pigmentosa GTPase regulator) have also been reported.3,4 Clinical phenotype of CGKD is related to the size of deletion and involved genes and the variability of clinical symptoms makes the diagnosis difficult. The majority of reported patients with CGKD exhibit salt-wasting dehydration during the neonatal period and childhood. Developmental delay (DD) is also frequently observed in males with Xp21 deletions, particularly when the deletion extends proximally to involve the DMD or when larger deletions extend distally to encompass both the IL1RAPL1 and DMD. 3
Here, we report the case of 2 male siblings with GKD, DMD and X-linked AHC as part of the CGS Xp2.1. The elder sibling presented with atypical features of congenital adrenal hypoplasia (CAH), leading to a delayed diagnosis. Whole exome sequencing (WES) enabled the diagnosis of CGKD, which highlights the importance of genetic testing in infants with atypical adrenal insufficiency and developmental delay. The younger sibling was diagnosed at birth, taking into account the family history, clinical and biochemical findings. As Infants with CGKD have a poor prognosis, early identification facilitates timely intervention and appropriate management.
Case Presentation
A 13-month-old male infant was referred for genetic evaluation due to metabolic abnormalities and developmental delay (DD). He was born after 37 weeks of gestation from first pregnancy with birth weight 3350 g (75th percentile) and length 52 cm (75th percentile). The pregnancy and delivery were uneventful. The patient was the first child of a young Caucasian nonconsanguineous couple. At birth, physical examination revealed hypotonia, hypospadias (Figure 1), scrotal hyperpigmentation and thickened penis with normal testicular volume (1 ml on each side). Soon after birth, the patient gradually developed lethargy and vomiting which required admission to neonatal intensive care unit (NICU). Laboratory tests revealed hyperkalemia and hyponatremia. The 17-hydroxyprogesterone (17-OHP) level was 1.699 ng/ml (normal range, 0.13-1.13). Based on the laboratory findings and scrotal hyperpigmentation hydrocortisone (5 mg administered intravenously every 8 hours) and fludrocortisone (0.1 mg taken orally once daily) were started on a presumed diagnosis of congenital adrenal hyperplasia (CAH), which stabilized his condition.
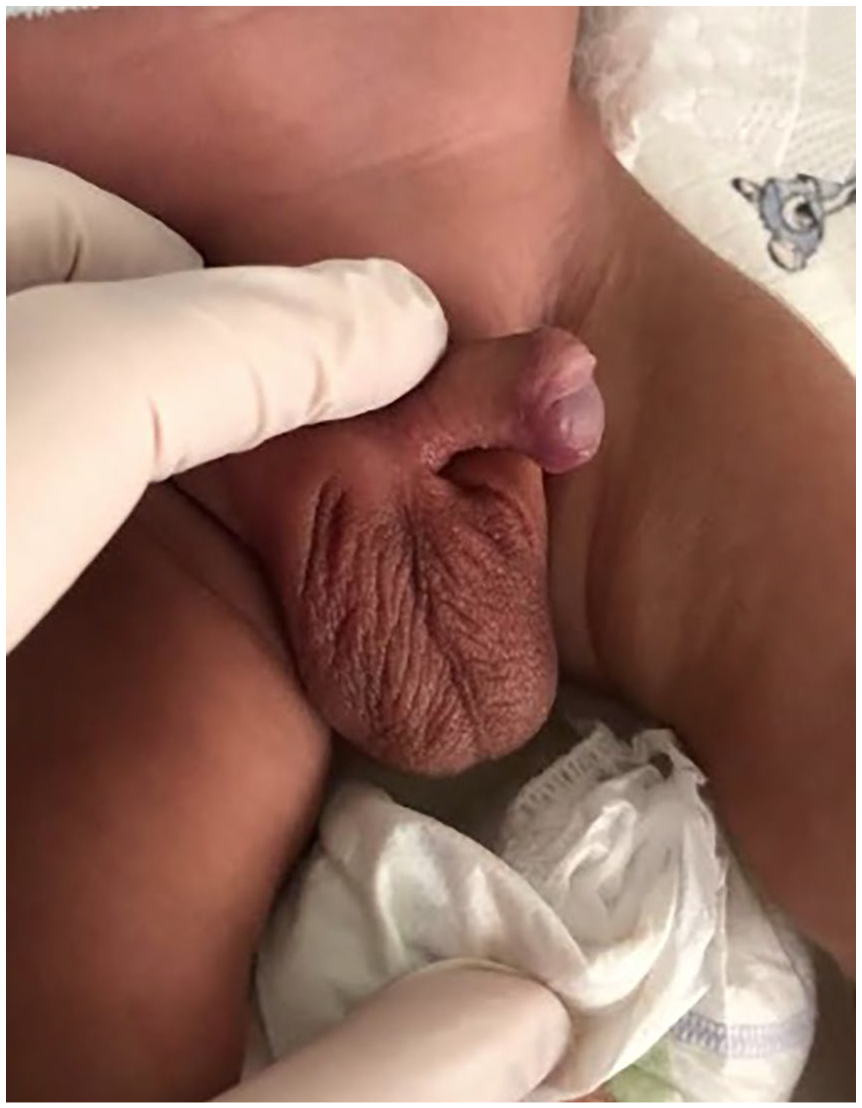
Photograph of the patient at 13 months of age showing hypospadias.
By time of physical examination at 13 months, his weigh was 7200 g (below the 3rd percentile) and length 68 cm (below the 3rd percentile). Clinical findings included hypospadias (Figure 1), scrotal hyperpigmentation, axial hypotonia, and dysmorphic facial features including high forehead, frontal bossing, rounded palpebral fissures, long philtrum, and downturned mouth. His developmental milestones were delayed, he could sit independently at 13 months but was unable to stand or walk independently. Intellectual development was also delayed: he had limited engagement in exploratory play, minimal verbal communication, and a lack of responsiveness to social interactions compared to age-matched peers. Upon neurological examination the patient appeared alert and attentive to surroundings. Cranial nerve examination and deep tendon reflexes were normal. Laboratory findings showed: potassium 4.08 mmol/l (normal range, 3.5-6.1), sodium 141 mmol/l (normal range, 133-142), low adrenocorticotropic hormone (ACTH) 4.24 ng/l (normal range, 4.7-48.8), low 17-OHP 0.31 μg/l (normal range, 0.8-2.0), low aldosterone 14.5 ng/l (normal range, 24-250), low dehydroepiandrosterone sulfate (DHEA-S) 0.01 μml/l (normal range, 0.09-3.4), normal free testosterone 0.32 to 01 μm/l (normal range, 0.31-1.3), renin, direct 2.36 ng/l (normal range, 1.7-23.9), aldosterone/direct renin concentration ratio 6.14 (normal <19), low fasting blood glucose level 60 mg/dl (normal range, 70-100). The urinary organic acid (UOA) profile was not assessed. Ultrasound imagining revealed normal abdominal and kidney findings, normal cardiac function, scrotal location and normal structure of the testicles. Routine G-banded karyotype analysis showed a normal male karyotype (46,XY). Whole exome sequencing (WES; CentoXome® Solo, Centogene, Rostock, Germany) identified an interstitial loss of 6.6 Mb involving the Xp21.3p21.1 chromosomal region (seq[GRCh37] Xp21.3p21.1(27478915_34150395)x0, size 6671 kb) classified as pathogenic based on American College of Medical Genetics (ACMG) criteria. 5 Deletion included the following genes: CXorf21, DCAF8L1, DCAF8L2, DMD, FAM47A, FTHL17, GK, IL1RAPL1, MAGEB1, MAGEB10, MAGEB2, MAGEB3, MAGEB4, MIR3915, MIR4666B, MIR548F5, MIR6134, NR0B1, PPP4R3C, TAB3. Targeted nucleotides covered ⩾20× 99.48%.
Based on clinical and genetic investigations the diagnosis of CGKD was made. Retrospectively, serum creatine kinase (CK) and fasting serum triglyceride (TG) levels were measured. CK was elevated—600 U/l (normal < 228), as well as TG—2.94 mmol/l (normal < 2.26). Fasting serum cholesterol was normal. Liver enzymes were elevated: alanine aminotransferase (ALT)—293 U/l (normal range, 0-35) and aspartate aminotransferase (AST)—358 U/l (normal range, 0-40). The patient frequently experienced episodes of low blood glucose, with glycemia levels of 50 to 60 mg/dl noted during routine laboratory evaluations. The parents reported episodes of irritability, which improved quickly after food intake.
At the time of diagnosis, the patient’s mother was pregnant with her second male child. He was born term and presented with similar clinical and biochemical findings as his older brother including electrolyte abnormalities, elevated CK and liver transaminases. WES revealed the same deletion as in his older brother. He received hydrocortisone and fludrocortisone replacement therapy following birth. Maternal testing confirmed her carrier status.
Currently, both patients are being managed with steroids and a diet of frequent carbohydrate-rich meals. The siblings are under continuous follow-up with a multidisciplinary team consisting of genetic specialists, metabolic dietitians, developmental pediatricians, speech therapists, physical therapists, endocrinologists, and neuromuscular specialists.
Discussion
Xp21 CGD syndrome is a rare genetic disorder resulting from the deletion of a chromosomal fragment in the Xp21 region that contains the critical GK locus. CGKD is characterized by significant variability in clinical presentations, making the diagnosis challenging. In the older sibling, the initial presentation with vomiting, poor weight gain, and hyperpigmentation led to a misdiagnosis of CAH. However, the diagnosis of CGKD allowed for more accurate management and genetic counseling for the family. The identification of the condition in the younger sibling allowed for earlier intervention and less complicated management.
Patients with atypical presentations of CAH should be assessed for the possibility of X-linked adrenal hypoplasia congenita. Early diagnosis, along with a multidisciplinary management strategy, is essential for optimizing patient outcomes. Once the diagnosis is confirmed, further screening for GKD and DMD is recommended. Early identification of GKD facilitates the implementation of comprehensive medical care, positively influencing the patient’s development and quality of life.6,7
In our case, WES identified an interstitial loss of 6.6 Mb involving the Xp21.3p21.1 chromosomal region. Deletion included GK, DMD, NR0B1, and IL1RAPL1. To our knowledge, the deletion of this specific size has not been reported; however, deletions involving all of these genes have been documented. 3
IL1RAPL belongs to the interleukin-1 receptor family and encodes protein which is expressed in brain and is involved in neurite outgrowth and functioning. Deletions in IL1RAPL1 have been reported in association with ID and autism spectrum disorder. 3 However, ID may occur without loss of IL1RAPL in the context of DMD-associated phenotype.
Females with CGKD are generally asymptomatic carriers; however, those with deletions may exhibit symptoms if skewed X-chromosome inactivation occurs. In the presented case, the mother was asymptomatic.
The treatment of CGKD focuses on dietary management, emphasizing regular intake of carbohydrate-rich meals, especially during illness or physical exertion. 7
Presented case also highlights the necessity of a multidisciplinary approach in managing CGKD, given its involvement of multiple systems, including metabolic, endocrine, neuromuscular, and developmental aspects.
Conclusion
In conclusion, the presented case demonstrates the complexity of diagnosing CGKD and reinforces the value of early genetic testing, comprehensive multidisciplinary care, and genetic counseling in managing this condition. The atypical presentation in the elder sibling emphasizes the need to consider CGKD in infants presenting with adrenal insufficiency and DD. WES proved essential for achieving an accurate diagnosis, while a multidisciplinary approach remains pivotal in addressing the complex and diverse clinical manifestations of CGKD.