
Abstract
Background:
Ataxia telangiectasia (A-T) is an uncommon autosomal recessive disorder, affecting 1 to 2 individuals per 100 000 live births. It results from mutations in the ATM gene. Patients typically present with progressive cerebellar ataxia, oculocutaneous telangiectasia, recurrent sinopulmonary infection, and predisposition to malignancies.
Case presentation:
This is a 10 year-old boy with recurrent chest infections and progressive gait imbalance since the age of 4, accompanied by ocular telangiectasia. Laboratory investigations revealed elevated serum alpha-fetoprotein (AFP) and hypogammaglobulinemia. Brain MRI showed cerebellar atrophy, and chest CT revealed pulmonary consolidation. These findings, together with clinical features, confirmed the diagnosis of A-T.
Clinical discussion:
Also referred to as Louis-Bar Syndrome, Ataxia-telangiectasia (A-T) is a multisystem genetic disorder characterized by a wide range of clinical manifestations. Diagnosis is based on a synthesis of clinical assessments and laboratory results, with genetic testing serving as the definitive method for confirmation. Management strategies are predominantly symptomatic and supportive, emphasizing immunoglobulin replacement therapy, immunization protocols, antibiotic administration for infection prevention, and vigilant surveillance for malignancies.
Conclusion:
This case emphasizes the importance of considering A-T in children with recurrent chest infections and neurological symptoms. Early diagnosis facilitates timely supportive care, including immunization, pulmonary management, malignancy surveillance, and genetic counseling for families.
Introduction
Ataxia telangiectasia (A-T), or Louis-Bar syndrome, is a rare autosomal recessive disorder affecting approximately 1 to 2 per 100 000 live births. 1 It results from mutations in the ATM gene on chromosome 11q22-q23, which encodes a kinase crucial for DNA repair and cell cycle control. 2 Dysfunction of this gene leads to a multisystem disorder characterized by progressive cerebellar ataxia, oculocutaneous telangiectasia, immunodeficiency, and increased cancer susceptibility. 3 Patients typically present with gait ataxia, recurrent respiratory infections, and ocular telangiectasia, while laboratory findings show elevated alpha-fetoprotein and reduced immunoglobulin levels. 4 MRI often demonstrates cerebellar atrophy, and genetic testing confirms the diagnosis. 5 There is no curative therapy; management focuses on infection prevention, immunoglobulin replacement, cancer surveillance, and supportive rehabilitation. Prognosis remains poor, with most patients becoming wheelchair-bound in adolescence.6,7 We report a 10-year-old boy with recurrent chest infections and neurological symptoms diagnosed with A-T—the first documented case in Yemen.
Case Presentation
A 10-year-old boy presented to the pulmonology clinic with productive cough, fever, dyspnea, and nasal congestion. The symptoms had a gradual onset and worsened over several days. His mother reported recurrent lower respiratory tract infections diagnosed as pneumonia since the age of 4, with transient improvement between episodes. He also had recurrent otitis media with purulent ear discharge.
Neurologically, the child had progressive gait imbalance and difficulty walking since age 4, gradually requiring assistance. Speech became slurred and effortful, with reduced articulation and mild dysphagia. There was a history of parental consanguinity, but no similar family illness (Figure 1).
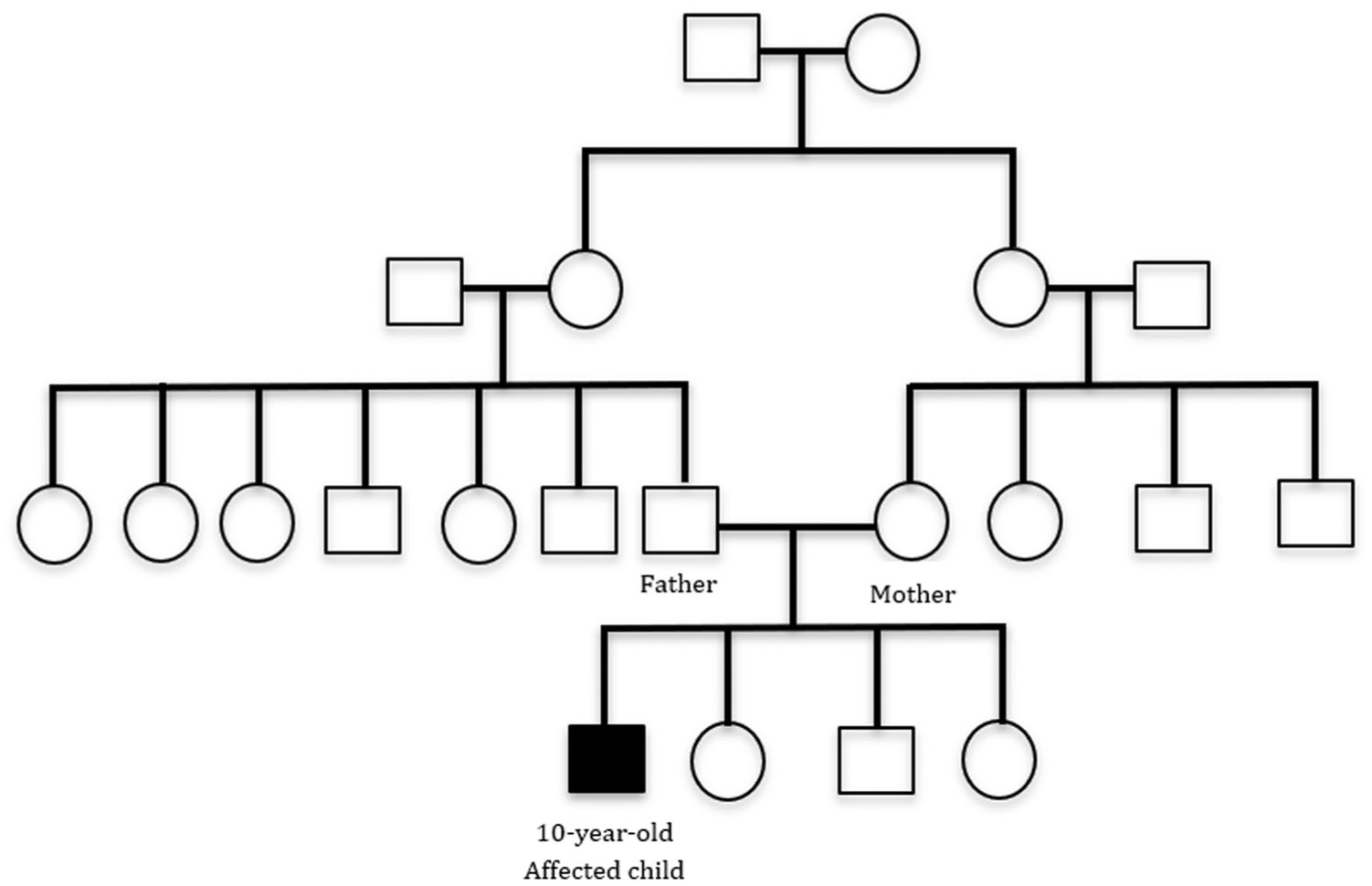
Pedigree chart of a 10-year-old boy showing a history of consanguineous marriage in the family.
On general examination, he was alert and oriented (GCS 15/15) but appeared underweight. Chest auscultation revealed inspiratory crepitations over the right lower lung zone. Neurological examination showed broad-based ataxic gait, dysarthria, dysdiadochokinesia, and positive cerebellar signs on finger-to-nose, finger-to-finger, and heel-to-shin testing. Romberg’s sign was positive. Ocular examination revealed oculomotor apraxia, horizontal nystagmus, and prominent bulbar conjunctival telangiectasias (Figure 2). Cranial nerve function was otherwise intact, and there was no sensory loss, extrapyramidal rigidity, or peripheral neuropathy.
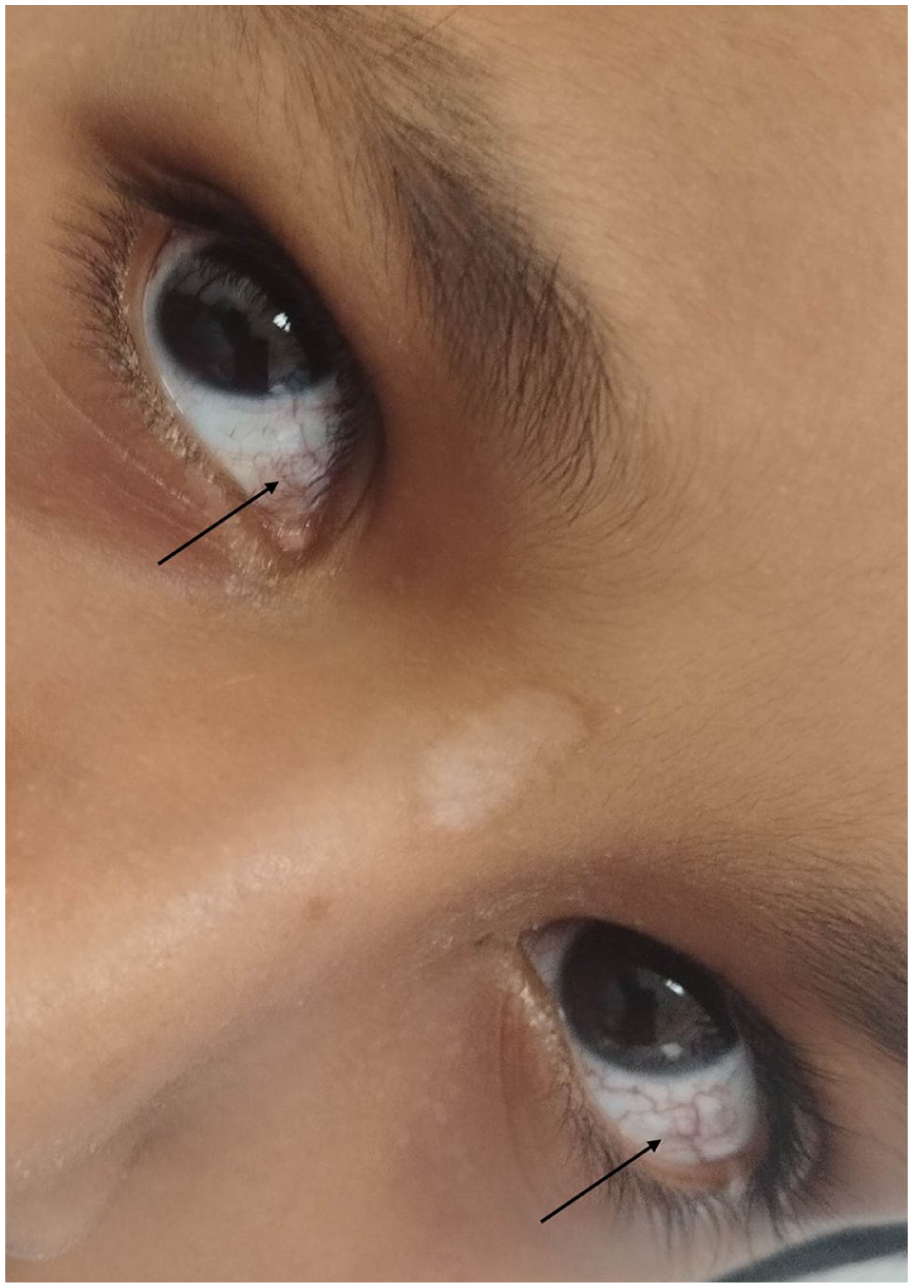
Bilateral ocular telangiectasia (black arrow) in a 10-year-old boy.
Laboratory results showed leukocytosis (18.8 × 103/µL) with neutrophilia, and elevated CRP (81 mg/L). Immunological evaluation revealed markedly reduced IgA (25 mg/dL), IgG (39 mg/dL), and IgE (1 IU/mL), with normal IgM (149 mg/dL). Serum alpha-fetoprotein (AFP) was significantly elevated at 201 ng/mL. Sputum culture grew Staphylococcus aureus. Chest CT demonstrated right lower-lobe consolidation with air bronchogram (Figure 3), and brain MRI revealed cerebellar atrophy (Figure 4).

Chest CT showing consolidation in the anterior right lower lobe with an air bronchogram (blue arrow).
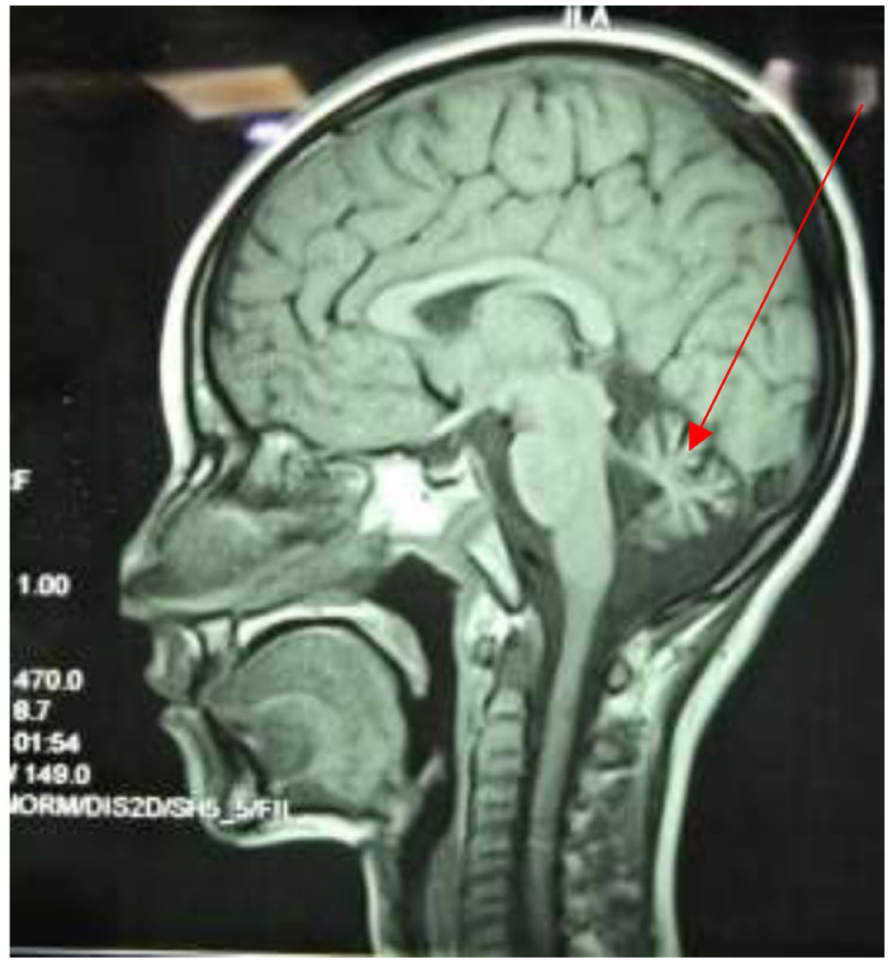
Cerebellar atrophy in T1-weighted sagittal cerebral magnetic resonance imaging (red arrow).
Based on the characteristic clinical features, elevated AFP, cerebellar atrophy, and immunoglobulin deficiency, a diagnosis of
The patient received antibiotic therapy for pneumonia and was started on intravenous immunoglobulin (IVIG) replacement. Influenza vaccination and speech therapy were advised. The family was counseled regarding the progressive nature of A-T, genetic implications, and the need for multidisciplinary follow-up with pulmonology, neurology, immunology, and rehabilitation services. The patient remains under regular follow-up and is clinically stable with ongoing supportive management.
Discussion
A-T is a rare autosomal recessive neurodegenerative disorder that affects about 1 to 2.4 per 100 000 live births globally. 8 It is caused by mutations in the ATM gene. 9 This condition is characterized by a combination of neurological, immunological, and systemic abnormalities, and typically manifests with progressive cerebellar ataxia, telangiectasia, recurrent infections, and an increased predisposition to malignancies. Symptoms usually manifest in early childhood, with ataxia being the earliest and most common feature, typically presenting within the first year of life. However, normal motor development between the ages of 2 to 5 years may mask the gradual progression of A-T, often leading to delayed diagnosis or misdiagnosis, as 60% of the patients are initially diagnosed with ataxic cerebral palsy.10,11 Oculocutaneous telangiectasias, the second hallmark of AT, usually manifest between the ages of 3 and 6 years, primarily affecting the bulbar conjunctiva, eyelids, and skin folds. However, telangiectasias may not be present in all patients, and their absence can contribute to delayed diagnosis.12,13 Additional neurological symptoms, such as oculomotor apraxia, nystagmus, dysarthria, myoclonic jerks, and tremors, can be present and significantly impair patients’ daily activities.14,15 The neurological issues in A-T are progressive, with many patients becoming wheelchair-bound by their teenage years. Furthermore, these neurological impairments can lead to pulmonary complications, which may cause recurrent respiratory infections.16,17 During the first 2 years of life, viral infections predominate, whereas in later childhood, common bacterial pathogens such as Hemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus, and Pseudomonas aeruginosa are more frequently encountered. These infections are closely linked to the severity of the humoral immunodeficiency, which justifies the use of intravenous immunoglobulin (IVIG) replacement therapy to reduce infection frequency and severity. 13 Additionally, aspiration pneumonia, resulting from dysphagia due to neurological deficits, and interstitial lung disease (ILD) are significant pulmonary manifestations in A-T. ILD affects approximately 25% of A-T patients, with a mean age of onset of 17.5 years. 18
The European Society for Immunodeficiencies (ESID) established diagnostic criteria for A-T, which include the presence of ataxia and at least 2 of the following: oculocutaneous telangiectasia, elevated AFP, chromosomal translocations involving chromosomes 7 and 14, or cerebellar atrophy observed through MRI. 19
The definitive diagnosis of A-T is confirmed by genetic testing for ATM gene mutations. In cases where genetic testing is unavailable, diagnosis can be made based on clinical symptoms, lab results, and imaging studies.
20
Key laboratory markers include elevated AFP, reduced immunoglobulin classes (particularly IgA, IgG2, and IgE), and lymphopenia, especially affecting T lymphocytes.20,21 Elevated AFP levels are a reliable and easily detectable biomarker, observed in over 90% of A-T cases.
22
MRI of the brain is an essential diagnostic tool, typically revealing cerebellar atrophy, cerebral white matter lesions, and enlarged fourth ventricles.
9
In the present case, differential diagnoses for progressive ataxia and immune dysfunction were carefully considered.
Conclusion
Ataxia telangiectasia (A-T) is a rare neuroimmunological disorder characterized by progressive cerebellar ataxia, oculocutaneous telangiectasia, and immune deficiency. Diagnosis relies on clinical presentation, elevated alpha-fetoprotein, characteristic MRI findings, and confirmation through genetic testing. Supportive multidisciplinary management—including intravenous immunoglobulin replacement, infection control, and rehabilitation—remains essential for improving patients’ quality of life.
This case represents the
Methods
The work has been reported in line with the SCARE criteria. 24
Footnotes
Acknowledgements
We hope to thank SMSR Team Lab. for their efforts and bringing our team together.
Ethical Considerations
Ethics approval is not required for case reports deemed not to constitute research at our institution
Consent for Publication
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Authors Contributions
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Research Registration Number
Not applicable because our article is a case report.
Guarantor
Anas Al-Kubati.