
Abstract
Kindler Syndrome (KS) is a rare, autosomal recessive genodermatosis caused by mutations in the FERMT1 gene, leading to skin fragility, blistering, photosensitivity, and progressive poikiloderma. We present a unique case of KS in a 6-year-old boy born to consanguineous parents, exhibiting uncommon dermatological, and systemic features. The patient developed multiple erythematous plaques, hemorrhagic crusting, and purulent discharge after birth, with a family history suggesting genetic predisposition. Uniquely, the patient presented with well-demarcated hyperpigmented macules on the abdomen, a feature rarely seen in KS, which adds to the phenotypic diversity of the condition. Additionally, the patient had extensive lanugo hair growth, nail dystrophy, and gingivitis, typical of KS, but without urinary or mucosal involvement, a departure from more classic presentations. The patient also presented with glucose intolerance, indicated by elevated glucose levels of 222 mg/dL, likely due to infection-induced metabolic dysregulation, which normalized after treatment. The differential diagnosis initially considered porphyria cutanea tarda (PCT) due to overlapping features like photosensitivity and skin fragility. However, laboratory findings, including normal liver function and the absence of specific PCT markers, effectively excluded PCT. Microbiological swabs from purulent discharge identified Staphylococcus aureus, which was sensitive to the prescribed antibiotics. Management focused on symptomatic relief with antibiotics, supportive care, and iron supplementation to address anemia caused by chronic skin erosions. The case highlights diagnostic challenges in resource-limited settings where genetic testing was unavailable. It underscores the need for heightened awareness of atypical KS manifestations, the importance of clinical evaluation and genetic counseling, and contributes to the expanding knowledge of KS, particularly in populations with consanguineous marriages.
Introduction
Kindler Syndrome (KS) is a rare autosomal recessive genodermatosis characterized by skin fragility, blistering, photosensitivity, and progressive poikiloderma. 1 It is an autosomal recessive disorder caused by mutations in the FERMT1 gene, which encodes the protein kindlin-1, located on chromosome 20p12.3, critical for integrin-mediated epidermal-dermal adhesion. 2 Since its initial description in 1954, 3 over 250 cases of KS have been reported worldwide, with the largest series including 26 patients from a Panamanian tribe. 4
Management strategies focus on symptomatic relief and prevention of complications, such as digital constriction bands, which may require surgical intervention. 5 While unique presentations of KS, such as lanugo hair and serious urinary complications, have been documented, 6 this rare genodermatosis continues to exhibit significant variability in its clinical manifestations. We present a distinct case of KS in a 6-year-old boy born to consanguineous parents, characterized by uncommon dermatological and systemic features, expanding the phenotypic spectrum of KS, and suggesting either a previously unrecognized overlap with neurocutaneous disorders or a novel variant of FERMT1-related genodermatoses requiring further molecular investigation. Furthermore, it emphasizes the importance of clinical evaluations and genetic counseling, particularly in resource-limited settings where genetic testing is not readily available. The case serves as a reminder of the complexities involved in diagnosing rare genodermatoses and the importance of considering atypical presentations when managing such conditions.
Case Presentation
A 6-year-old Pakistani male, born from a consanguineous marriage, presented with a chronic dermatological condition characterized by multiple erythematous plaques, hemorrhagic crusting, purulent discharge, and worsening symptoms over the past 10 days. This condition began at 3 days of age, initially manifesting as a blister on the left big toe, which evolved into widespread skin involvement. The patient’s family history reveals a deceased female sibling who had a similar skin condition and died at 6 months of age from severe systemic infections and aspiration pneumonia, suggesting a probable genetic factor as depicted in the Pedigree Diagram (Figure 1). Notably, there was no history of urinary problems or mucosal involvement.
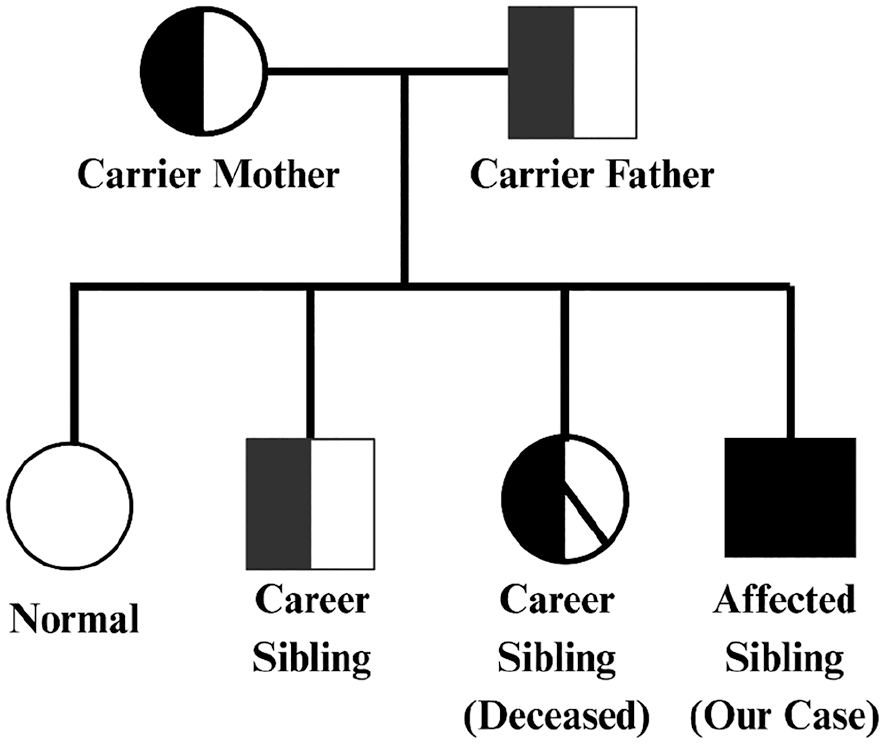
The family pedigree for KS illustrates the genetic inheritance pattern within the family. The full shading of a symbol represents an affected individual, in this case, the proband (the male patient in our case). Half shading indicates a carrier individual, such as the father, mother, and one of the siblings, who possesses 1 copy of the mutated gene but is not clinically affected. The diagonal shading through the symbol signifies a deceased individual, in this case, a carrier sibling who passed away. This pedigree highlights the autosomal recessive inheritance of KS, with the parents being carriers, leading to the potential for affected children.
At the time of presentation, the patient was febrile with chills and rigors. He also reported constipation but no swallowing difficulties. Dermatological examination revealed recurrent blistering, skin erosion, photosensitivity, fragile skin, and nail dystrophy with transverse ridges on hand nails and dystrophic toenails. Generalized hypo- and hyperpigmentation with skin atrophy were also observed (Figure 2). The patient’s facial findings included reticular hyperpigmentation and telangiectasias (Figure 1). Oral examination showed mild erosions and gingivitis (Figure 2). On further inspection, there was widespread growth of lanugo hairs on the face, legs, and neck. Upon further examination, there was extensive growth of lanugo hairs on the face, legs, and neck. This constellation of symptoms is classic for KS, a diagnosis that was made based on the patient’s history, clinical examination findings, and family history.

Clinical findings in a 6-year-old patient with Kindler syndrome: (a) hyperkeratotic plaques and cigarette paper-like wrinkling on the dorsum of hands, (b) atrophy of skin with hypo-hyperpigmented patches—poikiloderma, (c) visible area of hypo-hyperpigmentation with crusted plaques secondary to skin blistering, and (d) gingivitis with widely spaced and loss of teeth.
Notably, the patient presented with well-demarcated hyperpigmented macules localized on the abdomen, which were stable in size and shape. This finding is rarely observed in KS patients (Figure 3).
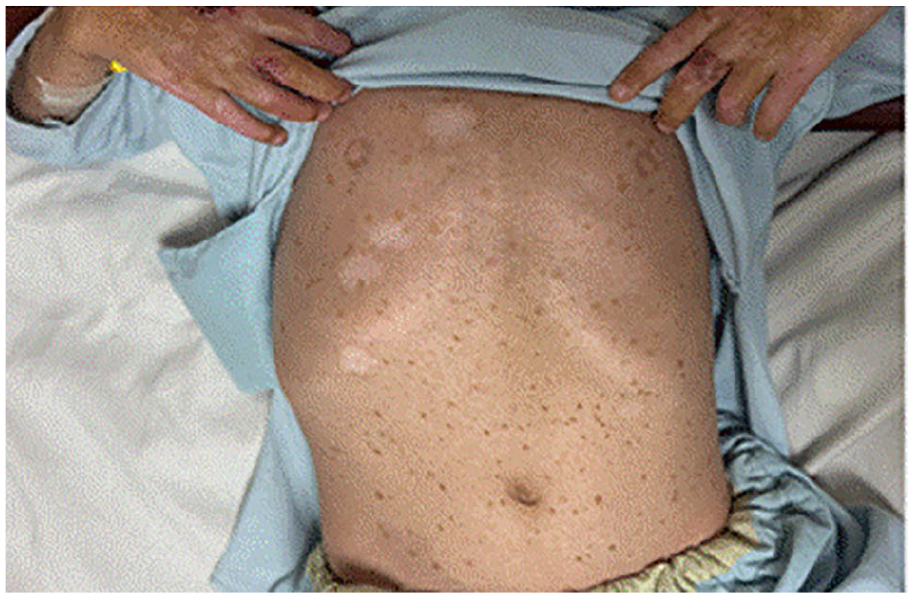
Well-demarcated hyperpigmented macules on the abdomen—a rare dermatological finding in Kindler syndrome.
The patient had previously been admitted for similar symptoms, which were treated with topical and intravenous antibiotics, normal saline washes, and soaks, leading to complete resolution between episodes, but recurrence occurred despite effective treatment. Laboratory assessments showed an elevated white blood cell count (16,000/µL) and microcytic anemia (MCV 67 fL), classified as iron deficiency anemia. The alkaline phosphatase (ALP) level was 281 U/L, which is within the age-specific reference range for a 6-year-old (273.47-871.44 U/L). However, due to the patient’s complex clinical presentation, further biochemical investigations were warranted. Additional tests, including serum electrolytes and liver function tests, were performed and found to be within normal limits, ruling out primary hepatic pathology. The patient exhibited elevated random glucose levels of 222 mg/dL, likely attributed to infection-induced metabolic dysregulation and a cytokine-driven stress response.
Our clinical evaluation included consideration of porphyria cutanea tarda (PCT) as a differential diagnosis due to shared features with KS, such as photosensitivity and skin fragility. However, the absence of key PCT clinical markers combined with diagnostic laboratory parameters effectively excluded PCT. The patient’s complete blood count showed leukocytosis (WBC 16 × 109/L) with neutrophilia (80.9%), consistent with secondary bacterial infection rather than a metabolic disorder. Liver function tests were normal, with ALT/GPT at 14.5 U/L and total bilirubin at 0.17 mg/dL, both well within normal ranges. Urinalysis revealed no urobilinogen or bilirubin with a normal urinary pH of 6 and an absence of pink-colored urine, which would be expected in PCT. The comprehensive metabolic panel showed no evidence of hepatic injury, and negative viral serologies (HBsAg, HCV, HIV) excluded triggering factors commonly associated with PCT. Figure 4 shows a timeline of significant events during the patient’s treatment course.

Key events in the patient’s clinical course.
Intravenous (IV) amoxicillin-clavulanic acid (300 mg twice daily, increased to 600 mg due to worsening symptoms) was administered. Topical treatment with fusidic acid ointment 2% was applied twice daily for localized skin infections, and normal saline soaks were administered thrice daily to manage wounds. Syrup Vidalyn (5 mL daily) was prescribed for constipation and nutritional rehabilitation. Microbiological cultures of purulent discharge from erythematous plaques identified Staphylococcus aureus, sensitive to amoxicillin-clavulanic acid and Fusidic acid, supporting the therapeutic regimen.
Genetic testing using Sanger sequencing, antigen mapping, and transmission electron microscopy was advised for a detailed examination of cellular and tissue structures. However, due to resource constraints, genetic testing could not be conducted, and the diagnosis was relied upon based on the clinical manifestations. Genetic counseling was provided to the family as a consanguinity-related inheritance pattern was suspected.
Follow-up assessments indicated significant improvement after antibiotic therapy, with hemoglobin levels increasing from 9 to 11 g/dL following iron supplementation and glucose levels normalizing to 98 mg/dL. Treatment adherence was monitored through clinical observations and caregiver reports. Minimal growth of commensal flora suggested effective antibiotic targeting, and therapy was continued without adjustment. Post-treatment swabs showed clearance of pathogenic bacteria, aligning with clinical resolution of purulent discharge. The patient responded to the treatment with minimal side effects. Fever and shivering were managed with supportive care and adjustments to the antibiotic regimen. At 6 months, the patient’s well-demarcated abdominal macules showed no progression, reinforcing their distinction from classic poikiloderma. Recurrent blistering was confined to trauma-prone areas (knees, elbows), managed with silicone-based dressings and strict photoprotection to mitigate UV-induced damage. A tailored dental regimen improved gingival health, while genetic counseling addressed familial recurrence risks, emphasizing prenatal testing options despite resource constraints.
Discussion
This case of a 6-year-old male with KS highlights the diagnostic complexities and challenges associated with this rare genetic disorder. KS, a form of epidermolysis bullosa (EB), often presents in infancy, as seen in this patient, with erythematous plaques, blistering, and progressive poikiloderma. The progression from localized blistering to widespread skin involvement underscores the progressive nature of the disease. Kindlin-1, the protein implicated in KS, is essential for integrin-mediated cell adhesion to the extracellular matrix (ECM), and its absence leads to skin fragility, blister formation, and photosensitivity due to impaired cellular response to UV radiation. 7 The patient’s symptoms of nail dystrophy and periodontal issues further align with the documented complications associated with KS, 1 while the absence of urinary problems and mucosal involvement complicated the case, as KS can sometimes involve mucosal surfaces, leading to complications such as oral ulcers, and periodontal disease. 8 The patient’s consanguinity and family history of a deceased sibling with similar symptoms further support the genetic predisposition of KS, which is inherited in an autosomal recessive manner. The genetic mutations associated with KS can be diverse, including nonsense mutations, deletions, and frameshift mutations, all of which lead to the loss of function of kindlin-1. 9 In our case, both parents are carriers of the mutated gene, which increases the likelihood of their offspring inheriting this autosomal recessive condition. 10 However, the absence of genetic testing, due to resource limitations, underscores a significant gap in confirming the diagnosis and understanding the specific mutation involved. This leads to the genetic counseling in our case being paramount. It was crucial to inform parents about the risks of recurrence in future pregnancies and the implications of the genetic findings for family members. Studies have shown that families with a history of KS may benefit from genetic counseling, which can help in making informed decisions regarding family planning and management of the condition. 11
KS is often misdiagnosed due to its similarities with various inherited blistering diseases, particularly other subtypes of EB, such as dystrophic, junctional, or simplex EB. The early presentation of blistering in infancy can lead to confusion, as these conditions share common features like skin fragility and blister formation. However, KS is characterized by a unique progression that includes photosensitivity and poikiloderma, which typically develop later in childhood. 12 In addition to EB, other conditions such as Rothmund-Thompson syndrome and dyskeratosis congenita also exhibit overlapping symptoms. Rothmund-Thompson syndrome includes poikiloderma and photosensitivity but is accompanied by distinctive features such as short stature and juvenile cataracts, which are absent in KS. 13 Similarly, dyskeratosis congenita presents with reticulated hyperpigmentation and nail dystrophy but lacks the blistering characteristic of KS. 14
The presence of well-demarcated, hyperpigmented macules on the abdomen in our patient is a noteworthy finding, as this presentation is rarely reported in KS. While KS is known to exhibit various cutaneous manifestations, including hyperpigmentation, the uniform shape, and size of the macules in our case are distinct from the more commonly observed mottled or reticulated pigmentation. Gupta et al 15 described irregular light-brown macules in KS, lacking sharp borders, while Ghosh et al 7 noted reticulated hyperpigmentation as part of progressive poikiloderma, not discrete macules. In contrast, the hyperpigmented macules in our patient were stable in size and shape, localized to the abdomen, and did not progress to poikiloderma, suggesting post-inflammatory changes rather than melanocytic hyperplasia. Although café-au-lait macules (CALMs) are associated with neurofibromatosis, the absence of systemic features like Lisch nodules or neurofibromas in our patient rules out this overlap. 16 This unique presentation underscores the phenotypic diversity of KS-related hyperpigmentation and highlights the importance of clinicopathological correlation to prevent misdiagnosis with other neurocutaneous syndromes.
The inclusion of PCT in our initial differential diagnosis was justified due to the overlap of clinical features with KS, particularly photosensitivity and skin fragility. However, several clinical and biochemical factors helped exclude PCT. First, our patient’s age (6 years) is atypical for PCT, which primarily affects adults aged 40 to 60. 17 Second, the laboratory profile showed normal liver function and no urinary porphyrin markers, both of which are hallmarks of PCT. 18 Additionally, PCT typically presents with elevated hepatic enzymes, abnormal urinary porphyrin excretion, and iron overload, none of which were detected in our patient. The characteristic mucosal fragility, acral blistering, and progressive poikiloderma, alongside the absence of hypertrichosis and sclerodermoid plaques, confirmed the diagnosis of KS, as it meets the established criteria for clinical diagnosis.19,20
The diagnostic challenges in our case extend beyond its characteristic dermatological manifestations and require careful interpretation of laboratory findings. The patient’s ALP level of 281 U/L initially raised concerns for hepatic or metabolic pathology. However, in pediatric patients, ALP follows a tetraphasic pattern, with peaks during infancy and puberty due to osteoblastic activity associated with skeletal growth. 21 Age-adjusted reference ranges for children (5-8 years: 273.47-871.44 U/L, 1-9 years: 100-420 U/L) place our patient’s value within normal physiological parameters for bone metabolism. 22 Thus, the elevated ALP level is more likely related to bone growth than hepatic pathology. ALP is not a specific inflammatory marker, and its elevation in dermatological conditions is often secondary to metabolic bone changes or nutritional deficiencies rather than cutaneous inflammation itself. 23 Additionally, the absence of cholestatic markers (eg, pruritus, jaundice), normal liver enzymes (ALT/AST), and normal bone growth obviates the need for hepatic imaging, as imaging is recommended only when ALP exceeds 1.5× the upper limit of normal, especially if accompanied by other abnormal liver function tests. 23
The patient’s microcytic anemia (MCV 67 fL) is primarily due to IDA, as demonstrated by the improvement in hemoglobin levels following iron supplementation. IDA in this case is likely a result of chronic iron loss from recurrent skin erosions associated with KS and impaired dietary iron absorption due to subclinical gastrointestinal microerosions. Additionally, anemia is exacerbated by chronic inflammation, with elevated levels of IL-6 and hepcidin in KS disrupting the body’s ability to recycle iron. 24 Inflammation from skin blistering further activates the TLR4/NF-κB pathway, a process in the body that activates inflammation in response to injury or infection, which can worsen conditions like anemia, and skin damage. 25 The increased energy demands from frequent wound healing also complicate the anemia, highlighting the need for continuous nutritional monitoring in KS management.
The elevated random glucose level of 222 mg/dL is likely a result of infection-related metabolic disturbances. The immune response, with elevated TNF-α and IL-6, impairs insulin function, while stress hormones like cortisol increase hepatic glucose production, contributing to elevated blood glucose. 26 Although KS may involve genetic disruptions affecting glucose regulation, the patient’s glucose levels normalized (98 mg/dL) after treatment, suggesting that the hyperglycemia was stress-induced rather than indicative of a chronic condition like diabetes. 27
The use of IV amoxicillin-clavulanate and topical Fusidic Acid 2% was guided by microbiological confirmation of Staphylococcus aureus infection in purulent plaques. This targeted therapy successfully controlled the acute infection, resolving fever and purulent discharge within 72 hours, highlighting the importance of culture-directed therapy in antibiotic stewardship for KS. 11 However, while such regimens manage acute infections, they do not address the underlying FERMT1 genetic defect that causes recurrent blistering and skin fragility. 28 Long-term surveillance is crucial, as chronic inflammation from recurrent infections can accelerate poikiloderma progression and increase the risk of squamous cell carcinoma (SCC). 29 Regular skin exams and patient education on sun protection, including UV-blocking clothing and topical sunscreens, are vital due to the photosensitivity inherent to KS.
This case highlights that in resource-constrained settings, a tiered diagnostic approach is essential. First-line assessments should include age-adjusted laboratory reference ranges and evaluations of nutritional status, particularly iron, and vitamin D levels. Second-line investigations may involve bone-specific ALP isoenzyme analysis and fasting glucose or HbA1c tests for persistent hyperglycemia. Genetic testing for FERMT1 mutations and EB subtype differentiation via immunofluorescence mapping should be prioritized in atypical cases. We emphasize the need for a multidisciplinary approach, integrating dermatological, nutritional, and metabolic perspectives to manage complex genodermatoses as observed in our case. Future research should focus on developing low-cost genetic screening tools and point-of-care nutritional assays to improve diagnostic accuracy in such cases.
Comparative Analysis of KS Cases From the Indo-Pak Subcontinent
The clinical patterns observed in KS across South Asian cases, from Pakistan and India (Table 1), consistently showcase photosensitivity, blistering, poikiloderma, and skin atrophy as defining dermatological features. Oral mucosal involvement, such as gingivitis and periodontitis, is a common manifestation, and systemic complications, including gastrointestinal issues, anemia, and photosensitivity-related eye symptoms, are frequently noted. Consanguinity is prevalent in many cases, reinforcing the autosomal recessive inheritance of the condition. In resource-limited settings, including our case, diagnosis often relies on clinical presentation due to the limited availability of genetic testing, which is often constrained by financial barriers. Our case adds a distinctive contribution to the existing literature, presenting well-demarcated hyperpigmented macules on the abdomen—an uncommon finding in KS cases. Additionally, while mucosal involvement in our patient was mild, the interplay of systemic and dermatological manifestations introduces a novel metabolic dimension to the disease, further expanding its phenotypic diversity. The family history of a deceased sibling with similar symptoms highlights the genetic underpinnings of the disorder, reinforcing the need for heightened awareness of KS’s complex and varied presentations.
Comparative clinical profile of Kindler Syndrome (KS) cases reported from the Indo-Pak subcontinent, highlighting demographic patterns, dermatological and systemic features, mucosal involvement, treatment approaches, and unique findings.

Source: The table underscores the clinical heterogeneity of KS, the frequent role of consanguinity, and the diagnostic and therapeutic challenges faced in resource-limited settings.
Conclusion
This case not only adds to the growing body of literature on KS but also emphasizes the need for comprehensive care strategies that address the dermatological, nutritional, and genetic aspects of the disorder. These patients should be referred to nutritionists for nutrition rehabilitation. The unique presentation of this case, coupled with the family’s genetic background, reinforces the critical role of genetic testing, and counseling in improving patient outcomes and guiding management strategies for similar cases in the future.
Footnotes
Acknowledgements
No acknowledgments to declare.
Ethical Considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent Statement to Publish
We have obtained the written informed consent of the patient’s parents for publication of their clinical details and imaging results.
Consent to Participate
Written informed consent was obtained from the patient’s parents for participation in this case report.
Author Contributions
Muhammad Aamir: Conceptualization, Investigation, Supervision, Writing-original Draft, Project Administration. Fahad Faizullah: Investigation, Conceptualization, Project Administration, Supervision, Writing-original Draft. Malik W.Z Khan: Investigation, Writing-original Draft, Writing-review and editing, Resources, Software. Touba Azeem: Writing-original Draft, Writing-review and editing, Resources, Software. Muhammad Awais Khan: Writing-review and editing, Resources, Software.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All data relevant to the patient have been included in this case report