
Abstract
Pena-Shokeir Syndrome (PSS) is a rare autosomal recessive disorder characterized by dysmorphic features, camptodactyly, arthrogryposis, intrauterine growth restriction, polyhydramnios, and pulmonary hypoplasia. Two types of this syndrome have been defined, differentiated by distinct clinical and genetic features. PSS is a potentially life-threatening condition, with most cases expected to be diagnosed prenatally via ultrasound. Genetic counseling is crucial to inform parents about recurrence risks and management strategies for future pregnancies. We report a case of PSS in a dichorionic diamniotic (DCDA) twin pregnancy. Despite normal prenatal ultrasounds, 1 twin was diagnosed postnatally with severe craniofacial anomalies, limb deformities, and pulmonary complications, consistent with PSS. In contrast, the second twin exhibited normal growth and development, with no anomalies identified. To the best of our knowledge, this is the third reported case of PSS in a twin pregnancy and the second involving a normal co-twin. This case aims to contribute to the existing literature by detailing the unique dysmorphic and clinical findings associated with PSS and emphasizing the diagnostic challenges in twin pregnancies.
Introduction
Fifty years have passed since Pena and Shokeir first described 2 rare autosomal recessive syndrome variants in 1974.
The first variant, known as Type 1, is a fetal akinesia or hypokinesia deformation sequence. It is usually characterized by multiple joint contractures, camptodactyly, pulmonary hypoplasia, and craniofacial anomalies, often accompanied by gestational complications such as polyhydramnios, intrauterine growth restriction, and reduced fetal movements. With a prevalence of 1 in 12 000 births,1,2 Type 1 frequently results in intrauterine death, neonatal demise, or early mortality due to lung hypoplasia.
The second variant, referred to as cerebro-oculo-facio-skeletal (COFS) syndrome or Type 2, is a rapidly progressive neurological disorder. This condition manifests with neurogenic arthrogryposis, microencephaly, intracerebral calcifications, infantile spasms, failure to thrive, and distinct facial features. Ophthalmological anomalies such as cataracts, microcornea, and optic nerve atrophy. Type 2 is typically fatal by the age of 5, underscoring its severe clinical course.
This case aims to shed light on the clinical, genetic, and pathogenic mechanisms underlying Pena-Shokeir Syndrome (PSS). We emphasize the importance of recognizing prenatal diagnostic features, which rely on typical ultrasonographic anomalies, while noting that a definitive diagnosis is usually confirmed postnatally.
Case Report
A female infant was born from a spontaneous dichorionic diamniotic (DCDA) twin pregnancy following normal prenatal ultrasounds. The pregnancy was uneventful, with no consanguinity or unusual exposures. Delivery was via cesarean section at 36 weeks of gestation due to maternal preeclampsia in a peripheral hospital. Apgar scores were 0, 8, and 9 at 10 minutes, requiring immediate resuscitation with ventilation, cardiac massage, and epinephrine at 20 minutes. The infant weighed 1830 g, measured 43 cm in length, and had a head circumference of 32 cm, all below the third percentile. The first twin was transferred to our hospital: a tertiary care center for further evaluation and management and the second was discharged in good health.
On clinical examination, the neonate exhibited distinctive craniofacial features, including a peaked upper lip, open mouth, coarse facial features, persistent facial edema, and dysplastic, low-set ears angled backward. Limb abnormalities included micromelia, arthrogryposis with limited limb extension, bilateral clinodactyly of the fifth fingers, bilateral adducted thumbs, limited hip adduction, and complete sural atrophy. Cranial anomalies revealed macrocephaly, a large anterior fontanelle, separated metopic sutures, an open posterior fontanelle, and both posterior and high-arched palates (Figures 1 and 2).
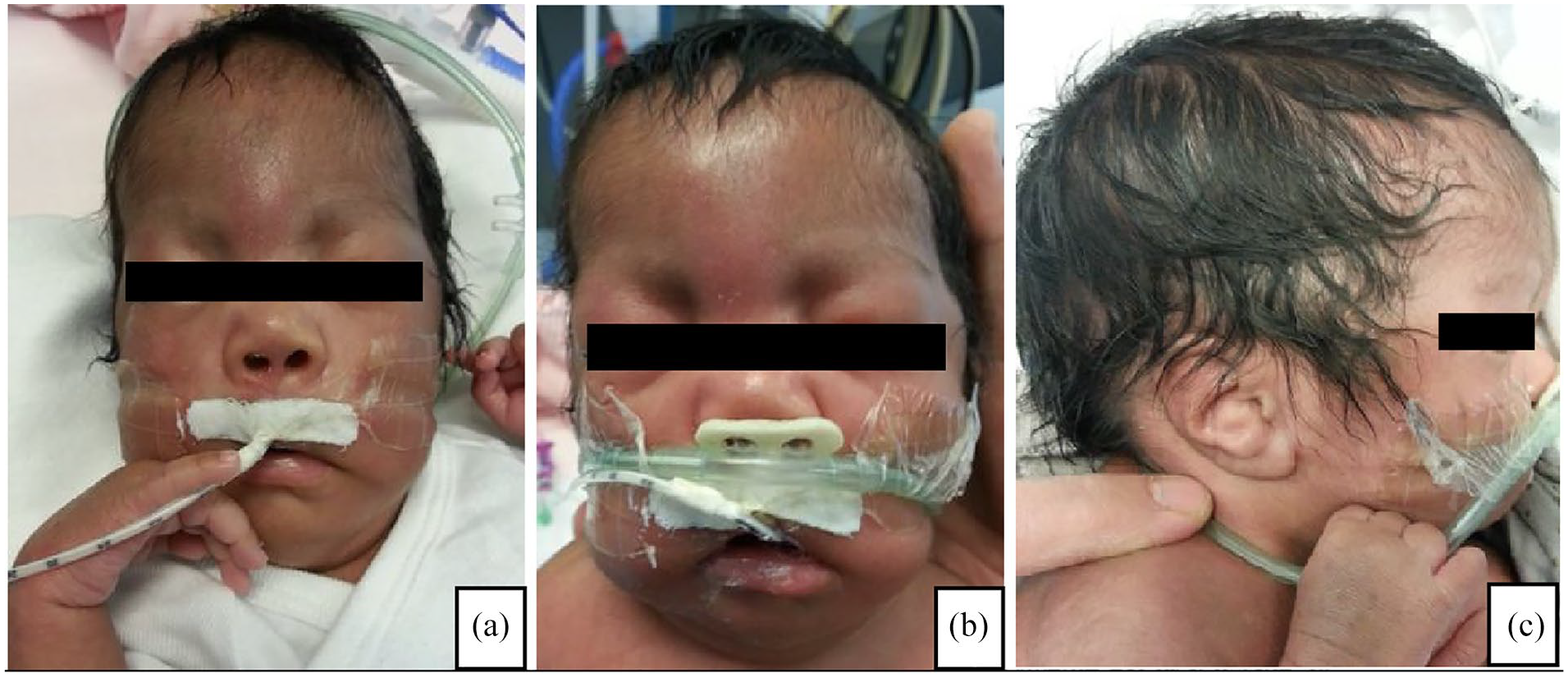
Pictures of the newborn with Pena-Shokeir syndrome revealing cranio-facial abnormalities: peaked upper lip (a and b) and low-set ears (c).
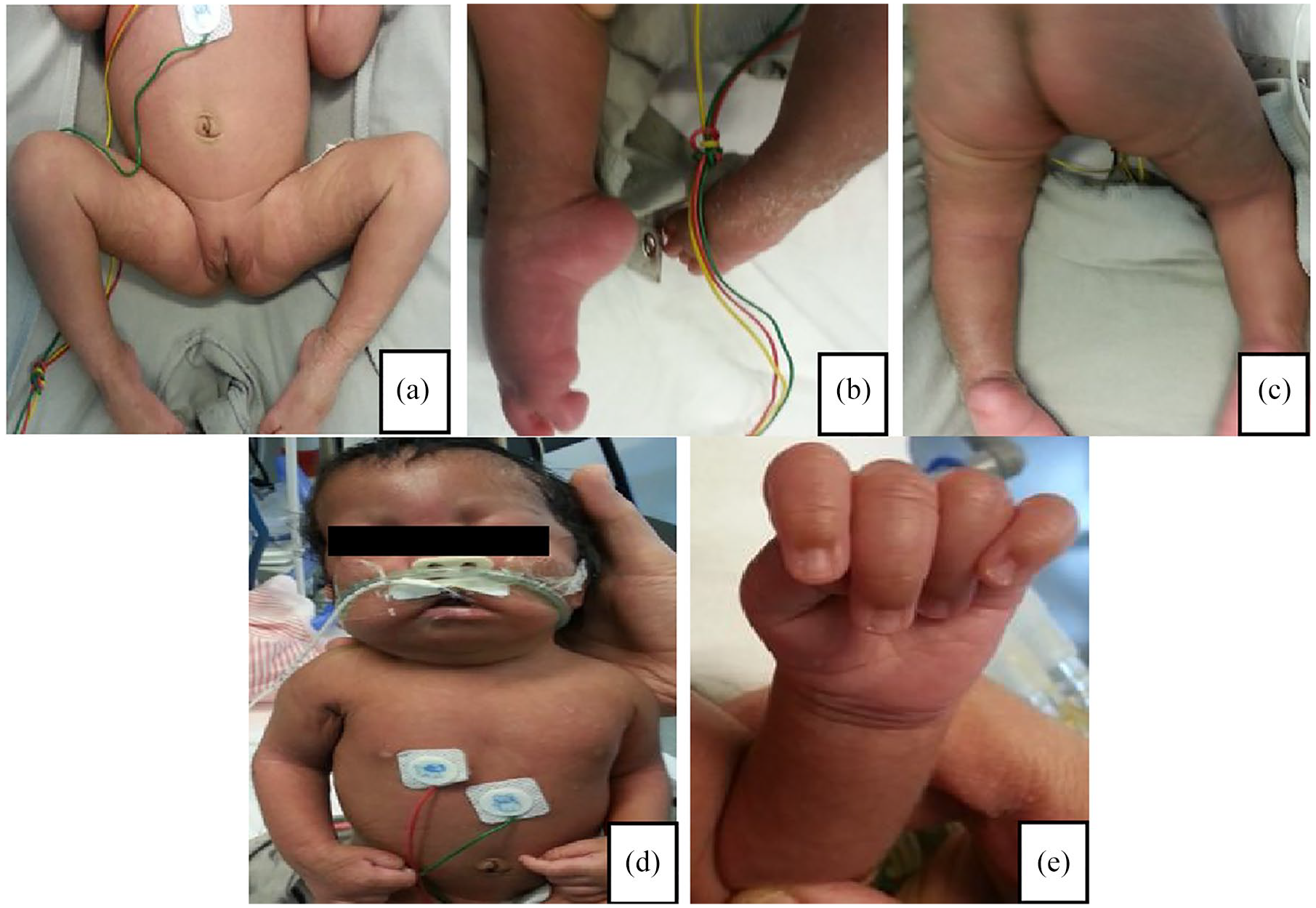
Pictures of the newborn with phenotypic features that include legs, arms and hand joint deformations: arthrogryposis (a-c) and bilateral adducted thumbs (d and e).
Neurologically, the infant displayed severe axial hypotonia with moderate peripheral hypotonia, fatigable eye contact, and absent vertical tracking. Weak horizontal tracking and sucking reflex were noted. Non-invasive ventilation in the prone position was required due to poor salivary clearance and recurrent upper airway congestion. Chest X-ray revealed right alveolar opacity. ENT endoscopy showed laryngeal insensitivity, vocal cord paralysis in the open position, salivary stasis, and suspected vagus nerve involvement, accompanied by absent cough and swallowing reflexes.
Investigations revealed a normal karyotype (46, XX), normal cranial ultrasound and MRI, and physiological patterns on EEG appropriate for gestational age but with poor asynchronous background activity. Eye fundus examination was normal, while auditory evoked potentials indicated bilateral deafness. Electroneuromyography of the limbs and face showed no abnormalities. Abdominal ultrasound and laboratory tests, including thyroid function, glucose, renal, and liver profiles, electrolytes, creatine kinase, lactate, very long-chain fatty acids, CDG syndrom, blood amino acid, urine organic acid chromatography, acylcarnitine profile were unremarkable.
These findings were consistent with Pena-Shokeir Syndrome (PSS). The diagnosis was communicated to the parents during a structured meeting with a multidisciplinary team, including a pediatrician, geneticist, and psychologist. The team prioritized clarity and compassion, ensuring that the information was conveyed in an accessible and supportive manner. The parents were informed about the clinical features of the syndrome, its poor prognosis, and the absence of available medical interventions for the affected twin. While they initially expressed grief, they gradually accepted the situation and found comfort in the health of the surviving twin.
The patient was unfortunately lost to follow-up and passed away from severe bronchiolitis at the age of 6 months.
Discussion
Pena-Shokeir syndrome is a rare lethal congenital disorder with an estimated incidence of 1/12 000.1,3,4 It was initially identified by Pena and Shokeir in 1974. Therefore, more than 100 cases have been published.
Autosomal-recessive is the most common mode of transmission. The risk reccurence in subsequent pregnancies is uncertain. However, it has been approximated to vary between 0% and 25%. 5
The cause of this condition isn’t clearly identified. The disorder of the neuromuscular system including: brain and spinal cord, motor neuron, neuromuscular junction, and neurotransmitter defects can explain all symptoms of the Pena-Shokeir syndrome. 6 This dysfunction leads to reduce fetal movements in the intrauterine period.
In recent observations, some women with myasthenia gravis 7 suggest that the pathogenesis happens at the neuromuscular transmission.
Maternal disease, metabolic and toxic factors could be implicated in some cases. 3
Mutations in RAPSN (Online Mendelian Inheritance in Man [OMIM] 601592), DOK7 (OMIM 610285), and MUSK (OMIM 601296) contribute to the development of PS by impairing interactions at the neuromuscular junction. Genetic testing wasn’t performed in this case due to financial limitations.
Autosomal dominant neuromuscular disorders may manifest as PSS when the genetic mutation occurs in a homozygous state. 8
Prenatal diagnosis can be achieved as early as 12 weeks. Prenatal ultrasonographic findings are intrauterine growth restriction, decreased fetal movements, short umbilical cord, joint contractures, pulmonary hypoplasia and polyhydramnios. Prenatal diagnosis was not performed in this case.
Another method to diagnose Pena-Shokeir syndrome is magnetic resonance imaging (MRI) is an other imaging method to diagnose Pena-shokeir syndrome. It is essentially requested when there is suspicion of fetal central nervous system anomalies.
If PSS had been identified earlier, it could have provided several benefits in terms of prenatal counseling and care planning. Early detection could have allowed the parents to better understand the prognosis, prepare emotionally, and make informed decisions about the management of the pregnancy. However, given the severe prognosis associated with PSS and the absence of curative interventions, the ultimate outcome for the affected twin would likely have remained unchanged.
This case highlights the importance of accessible and advanced prenatal diagnostic tools, especially in resource-limited settings, to enable earlier detection of rare syndromes.
Craniofacial anomalies include hypertelorism, micrognathia, apparent short neck, low set ears, and depressed nose tip.3,9 There are skeletal and joint anomalies: arthrogryposis, camptodactyly, ulnar deviation of hands, Equinovarus deformity/rocker-bottem feet, Kypho/scoliosis, webbing/pterygium, syndactyly, preaxial polydactyly.
Elias et al 10 described an infant with cerebellar hypoplasia. Dimmick et al 11 reported that PSS is associated with CNS damage involving cerebrum, cerebellum, thalamus, brainstem, corpus callosum, and spinal cord fibrosis. It results in hypotonia, infantile spasm, seizures, hemiparesis, hydrocephaly and mental retardation
Other cerebral, endocrine, gastrointestinal, genitourinary (cryptorchidism), and cardiovascular abnormalities have been reported.3,12 The neonate described by Almoshantaf et al 13 underscores the critical importance of early diagnosis and intensive management of the respiratory and cardiac manifestations such as the atrial septal defect and tricuspid valve insufficiency.
We consider that our case was compatible with PSS due to clinical features which especially distinguish PSS from other conditions and the karyotype was normal. Nevertheless, the antenatal ultrasound was unremarkable.
Chen reported 5 cases of Pena-Shokeir: 1 had earlier given birth to a 25 week gestation twins after a complicated pregnancy by hydramnios. They had joint contractures, club feet, and minimal neck webbing. They died because of hypoplasia lung. 14 Mindy revealed that the first twin of a twin pregnancy wasp remature but normal. The autopsy of the second twin showed arthrogryposis, pulmonary hypoplasia, muscle atrophy, flexion contractures ans abnormal facial features compatible with PSS. 15
Polyhydramnios results from absence of swallowing, craniofacial manifestations results from underdevelopment of facial muscles, multiple joints contractures results from absence of fetal movements and hypoplasia lung is associated with a loss of rythmic thoracic movements.
Differential diagnosis involves: Trisomy 18, mucopolysaccharidosis, Freeman sheldon syndrome, lethal multiple pterygium syndromes (LMPS), Potter syndrome, Neu-Laxova syndrome, restrective dermopathy, Larsen syndrome.
Pulmonary hypoplasia, a hallmark feature of PSS, is typically attributed to fetal akinesia and oligohydramnios, both of which are central to its pathophysiology. Although pulmonary hypoplasia also seen in Trisomy 18, is often accompanied by congenital malformations. In contrast, LMPS may present with less pronounced pulmonary hypoplasia, as the primary pathology is extensive webbing (pterygia) across joints, combined with generalized akinesia.
In PSS, contractures are severe and often involve adducted thumbs, fixed postures, and limited fetal movement. While inTrisomy 18, they tend to be less severe and are typically associated with other characteristic features, such as clenched fists and overlapping fingers. LMPS is characterized by joint contractures that are intrinsically linked to the presence of pterygia, a feature absent in PSS.
Additionally, PSS is frequently associated with facial abnormalities such as micrognathia and low-set ears, along with intrauterine growth restriction and fetal demise. Trisomy 18 is marked by systemic malformations, including cardiac defects, brain anomalies, and distinctive craniofacial features. LMPS is further set apart by findings such as cystic hygroma, hydrops fetalis, and extensive skin webbing, which are not observed in PSS.
This syndrome has been described in 2 variants: Type I and II. Most liveborns with Pena-Shokeir type I die in utero or during the first days of life. 16
Pena-Shokeir II syndrome is linked with longer survival. 17 Pena-Shokeir II syndrome is distinguished from PSS type I by absence of pulmonary hypoplasia, the presence of microcephaly and ocular findings (microphtalmia, cataracts).
Boesen PV described a case of a 9 year old girl with Pena shokeir who survived with an impaired intelligence.
Families should be notified about the diagnosis and prognosis by a multidisciplinary team including obstetricians, neonatologists, radiologists, geneticists, surgeons, and phyiotherapists. Pulmonary complication is the primary cause of mortality.
There is unfortunately no specific therapy available for individuals with Pena-Shokeir syndrome. They can receive lifesustaining and supportive treatment including invasive ventilation, cardiopulmonary resuscitation, anticonvulsive therapy, antibiotics, and inotropes/vasopressors.
Conclusion
PSS is a very rare lethal syndrome with poor prognosis. In our knowledge, this is the first case reported of a twin pregnancy in our country. Professionals should be aware of prenatal ultrasonic features to fulfill a definitive diagnosis after birth. Since it is an untreatable condition, appropriate treatment options and palliative care should be proposed to children. Their parents require detailed genetic counseling and early recognizing in further pregnancies.