
Abstract
This case report describes a 23-year-old male patient who presented with right chylothorax as the initial manifestation of a severe flare of systemic lupus erythematosus (SLE) and secondary Evans syndrome. Chylothorax and chylous ascites are rare features of SLE that can occur due to the accumulation of triglyceride-rich fluid in serous cavities. However, they have never been reported as the initial manifestation of a lupus flare. Evans syndrome is a rare disease characterized by autoimmune hemolytic anemia and immune thrombocytopenia, which can be secondary to SLE. The concomitant occurrence of both chylothorax and Evans syndrome in the setting of systemic lupus erythematosus has never been described, and the exact causative mechanisms of both entities are yet to be fully understood. In this report, we discuss our approach to this challenging case to broaden the understanding of the clinical manifestations of systemic lupus erythematosus. Our findings emphasize the importance of considering rare features of systemic lupus erythematosus and secondary diseases when evaluating patients with the disease.
Keywords
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that manifests as a complex, inflammatory disorder affecting multiple organs, including the kidneys, skin, central nervous system, and blood cells. The clinical presentation of SLE is diverse, making diagnosis challenging, and rare manifestations may occur. Among these, pericardial and pleural effusions are common, with reported incidences ranging from 12% to 56%. 1 However, chylous pleural effusion (CPE), characterized by the accumulation of triglyceride-rich fluid in the pleural space, is a rare complication of SLE with few reported cases. CPE can result from autoimmune diseases, hematological malignancies, or infections such as tuberculosis. 2 Rare causes include lymphatic overgrowth, as in Gorham-Stout disease. 3 Although the pathophysiology of CPE remains unclear, it is believed to be associated with lymphatic inflammation and antigen-antibody complex formation.
Inflammatory processes associated with systemic lupus erythematosus (SLE) can also affect the hematological system. Patients with SLE have a reported incidence of 5-10% for autoimmune hemolytic anemia (AIHA) and 20-40% for immune thrombocytopenia (ITP). 4 Although AIHA and ITP often coexist, sequential occurrence as part of a condition known as Evans syndrome (ES) is rare. ES can be caused by hematological malignancies, infections, primary immunodeficiencies, and mainly autoimmune diseases, with 16% of cases attributed to SLE. 5
This report describes a young patient who presented with several rare complications arising from SLE, all occurring simultaneously. This is the first case reported in English literature of a massive chylothorax as the initial manifestation of an SLE flare. Furthermore, this is the only case known to date that has presented with both chylothorax and Evans syndrome (ES). Due to the complex nature of the patient’s symptoms, a thorough diagnostic workup was necessary to rule out other potential causes, such as infections and malignancies. Recognizing these clinical findings as manifestations of SLE could aid in early diagnosis and appropriate management of these rare complications.
Case Report
A 23-year-old male patient came to the emergency department following a 1-week clinical course of general malaise, productive cough, fever, diarrhea, and abdominal pain.
At 16, he was diagnosed with systemic lupus erythematosus (SLE) due to arthritis, alopecia, acute cutaneous lupus, hypocomplementemia, and a positive antinuclear antibody titer. He also had a history of triple-positive antiphospholipid syndrome (APS) that resulted in multiple thromboembolic events such as deep vein thrombosis, massive splenic infarct, stroke, and myocardial infarction on separate occasions. The patient had been receiving treatment with Azathioprine (50 mg t.i.d), Hydroxychloroquine (200 mg daily), and Prednisolone (5 mg daily) but had discontinued his medication 20 days before the consultation and was not attending regular medical checkups.
Upon physical examination at admission, the patient exhibited tachycardia (110 beats per minute) and decreased breath sounds in the right lung during auscultation. Laboratory findings revealed mild normocytic anemia (hemoglobin: 11.8 g/dL), moderate thrombocytopenia (69 000 cells/uL), low complement levels (C3: 52 mg/dL and C4: 11.6 mg/dL), and elevated inflammation markers, including C-reactive protein (47 mg/l), erythrocyte sedimentation rate (129 mm/h), and ferritin (398 ng/mL). However, the white blood cell (WBC) count (4.6 cells/uL) and neutrophil count (3.1 cells/uL) were within the normal range, and no abnormal cell morphologies were observed on the peripheral blood smear at this point. The chest X-ray (CXR) showed a massive right pleural effusion (Figure 1, Panel A).

Upper Panel: Posteroanterior (A) and lateral (B) views of a chest X-ray taken upon hospital admission, revealing a radiolucent image in the right hemithorax indicative of a massive pleural effusion. The X-ray displayed a complete “white-out pattern,” indicating a substantial accumulation of fluid in the pleural space. Lower Panel: High-resolution chest computed tomography, (C) coronal view, and (D) axial view. The images exhibit chylothorax as a septated pleural effusion, with the fluid not following gravity due to the presence of fibrotic tissue. Additionally, reactive paratracheal nodules are observed.
Empirical antibiotics were initiated as bacterial pulmonary involvement could not be excluded. However, antibiotic therapy did not improve the patient’s condition. A full workup to exclude infectious and malignant etiologies was performed (Table 1). An abdominal ultrasound revealed no significant findings.
Laboratory results, including pleural fluid analysis, essential rheumatological tests, and the infectious panel.

These tests were critical in confirming the diagnosis and ruling out other possible diagnoses.
A total of 2400 cc of odorless milky fluid was obtained through a therapeutic thoracentesis. Fluid cultures showed negative results, and the cytochemical analysis revealed elevated triglyceride levels (287 mg/dL) with low cholesterol levels (<50 mg/dL), thus confirming the diagnosis of chylothorax (refer to Table 1). White blood cell differential in the fluid was not suggestive of infection. Cultures and molecular tests for Mycobacterium were negative, and the adenosine deaminase level was 19 U/L. No tumor cells were found in the fluid cytology.
At that time, the patient’s SLE activity scores showed high disease activity with a SLEDAI-2K score of 18 points, attributed to polyarthritis, inflammatory rash, alopecia, oral ulcers, pleural effusion, low complement levels, fever, positive anti-DNA autoantibodies, and thrombocytopenia. Thus, after excluding other etiologies and infection, the medical team initiated treatment with methylprednisolone (1 mg/kg dose). Following 3 days of treatment, the patient’s fever subsided, and the rash improved; however, the patient continued to experience arthritis and thrombocytopenia.
The patient’s anemia worsened during the following days, with hemoglobin level dropping to 7.5 g/dL 5 days after admission. No sources of bleeding were identified. Autoimmune hemolysis was confirmed through elevated levels of lactate dehydrogenase (593 U/L), hyperbilirubinemia (Total: 2.24 mg/dL, Direct: 0.04 mg/dL, Indirect: 2.2 mg/dL), a reticulocyte count of 5%, and low haptoglobin levels (37 mg/dL, reference range: 41-165 mg/dL). The direct Coombs test was positive for IgG. A new peripheral blood smear revealed anisocytosis, polychromatophilia, and elliptocytes. Notably, at this point the patient’s previously normal white blood cell count concomitantly fell to 1.4 cells/uL (neutrophil count: 1.0 cells/uL). A bone marrow aspiration ruled out hematopoietic malignancies, and other causes of thrombocytopenia, such as thrombotic thrombocytopenic purpura (TTP), were excluded (Plasmic score: 3, ADAMST-13: negative). The sequential development of immune thrombocytopenia (ITP), autoimmune hemolytic anemia (AIHA), and autoimmune neutropenia (AIN) was consistent with the diagnosis of secondary Evans syndrome. The patient did not fulfill the criteria for macrophage activating syndrome. Follow-up chest CT scan were obtained through this period revealing a persistent septated pleural effusion and enlarged reactive paratracheal nodules (Figure 1, Panel B).The pleural effusion was managed with open thoracostomy, but multiple subsequent thoracenteses were required to control the effusion, and none of them were initially successful.
Based on the presented findings and the patient’s persisting arthritis and life-threatening hematological symptoms, intravenous immunoglobulin (IVIG) treatment and immunosuppression with cyclophosphamide were initiated. AIHA and AIN resolution was achieved (toward the fourteenth day of admission), while the platelet count started to improve steadily. The evolution of hematological parameters through admission is better shown in Figure 2. Additionally, after administration of immunomodulatory management chylothorax started to improve modestly.
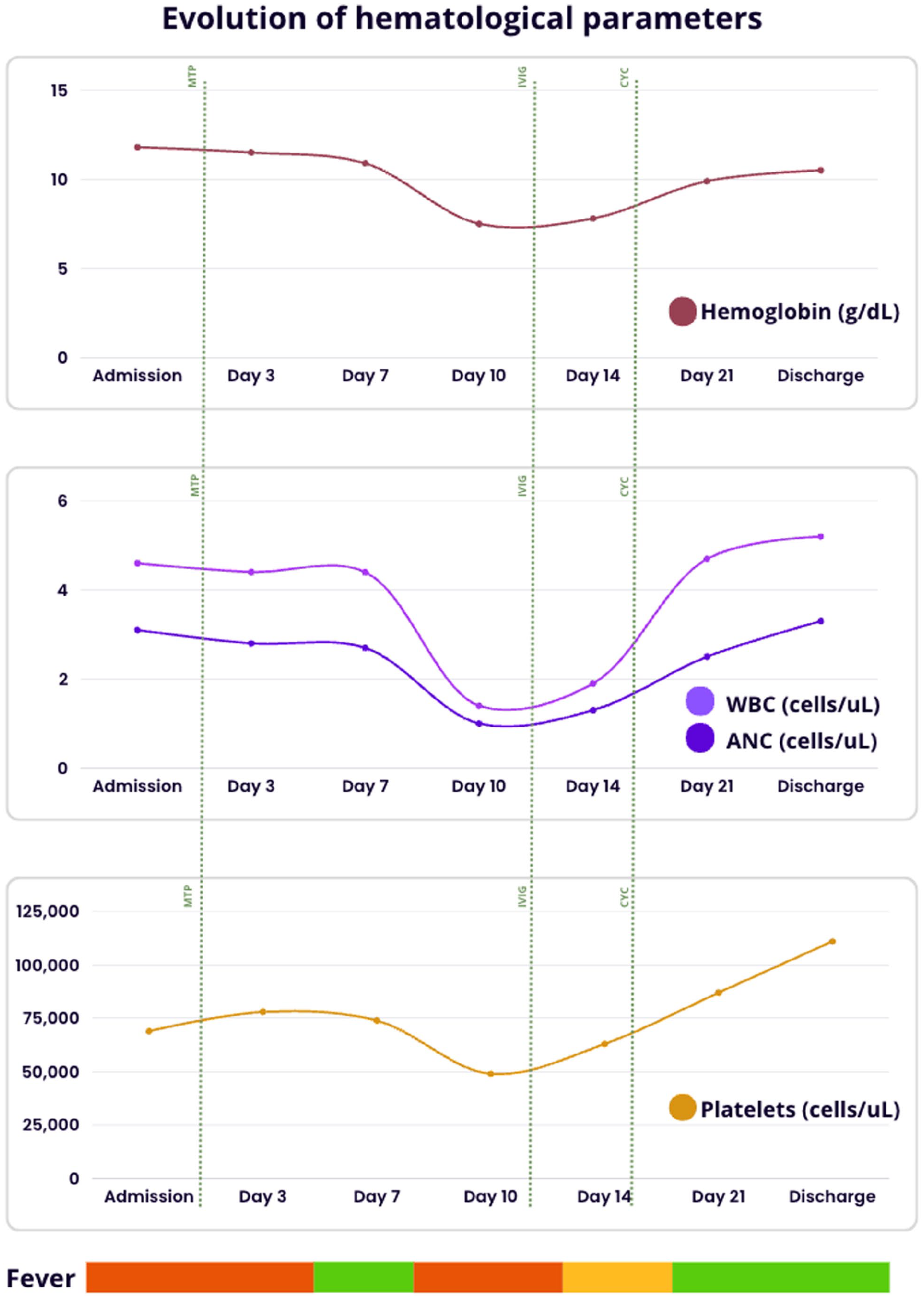
Evolution of hematological parameters during hospitalization, depicting hemoglobin levels in red, white blood cell count (WBC) and absolute neutrophil count (ANC) in purple, and platelet count in yellow. A decline in all 3 parameters is observed until day 10 of hospitalization, followed by improvement after the administration of immunomodulatory management. The association with fever is displayed in the lower section, with the green bar representing no fever, the red bar indicating constant and high-grade fever, and the yellow bar representing intermittent low-grade fever. The green dotted lines denote the administration of different treatments: MTP (methyl prednisolone), IVIG (intravenous immunoglobulin), CYC (cyclophosphamide).
Ultimately, the patient was discharged to continue with rheumatology follow-up. Six months later, imaging indicated only mild pleural effusion and the normalization of reactive nodules. The patient is currently taking azathioprine and prednisolone at a daily dose of 10 mg, and during the follow-up period of 18 months, no recurrence has been reported. The case timeline is better shown in Figure 3.

Case report timeline of the hospitalization happenings. The colored squares show medical interventions perform in each timeframe.
Discussion
Although pleural effusions are joint in SLE, chylothorax and chylous ascites are extremely rare, with only a few cases reported in English literature. 6 Among these cases, only 2 were from Western populations, one was Latin-American, and only 2 had (non-massive) chylothorax as the initial manifestation of SLE.7,8 The mechanisms by which SLE causes fluid accumulation have yet to be entirely understood. Inflammation of the lymphatic vessels may increase intraluminal lymphatic pressure, leading to fluid drainage into the serous cavities. Other theories suggest that reactive lymphadenopathies can cause secondary thoracic duct obstruction.
In the study by Zhang et al, chylous effusions were found in patients with low disease activity, which contrasts with our case where chylothorax occurred with a high SLEDAI-2K score (6.4 ± 2.1vs 18 points). 6 The time elapsed after diagnosis has also been recognized as an influencing factor in the presentation of chylous effusions. Two studies reported the occurrence of chylothorax at 120 and 20 to 120 months, respectively, after the diagnosis.9,10 Our patient shared this characteristic as he was diagnosed 86 months prior. However, more extensive studies are required to understand the risk conferred by these factors through multivariate regression analyses, as selection bias cannot be excluded with this sample size. A pleural triglyceride level >110 mg/dL with low pleural cholesterol levels is considered the gold standard for diagnosing CPE. Lymphoscintigraphy has been shown to provide additional anatomic information, but a precise diagnostic algorithm is still lacking.
Treatment strategies implemented were heterogeneous among reported cases, but most patients received corticosteroids as an initial treatment. Immunosuppression was frequently required and used heterogeneously, with some patients requiring surgical intervention. Interestingly, in the Zhang et al cohort, the decrease in SLE disease activity did not directly correlate with the resolution of CPE. 6 The observed refractoriness could be linked to lymphoscintigraphy findings of residual adhesions or thoracic duct obstructions that persist after SLE systemic inflammation’s resolution. 10 Surgical procedures may be needed in these cases and have shown better responses than medical therapy alone. In our case, medical treatment was ineffective in the short-term follow-up but achieved remission in the long term. Further studies are needed to clarify the preferred treatment, the differences between medical and surgical treatment, and the best intervention timing.
The development of secondary Evans Syndrome (ES) during the disease course was another notable finding in our case. Although hematological alterations are common in Systemic Lupus Erythematosus (SLE), ES is rare, occurring in only a slim percentage of cases. Additionally, overlapping autoimmune neutropenia is a rare complication of ES, seen in only 15-20% of cases. 11 A reported cohort of 26 patients with both SLE and ES demonstrated 2 noteworthy similarities to our case. 12 First, the development of secondary ES occurred in the context of severe multi-systemic involvement. Second, 34.6% of patients had other autoimmune diseases, with antiphospholipid syndrome (APS) being the most frequent (23%). The resolution of ES was proportional to the decrease in SLE disease activity. However, there are currently no specific guidelines for treating secondary ES. In observational studies, corticosteroids and IVIG have been used as treatment options, with 68% of patients having a partial response to corticosteroids. 11 Nevertheless, most patients experience relapses over time. Rituximab and bortezomib have been successful alternatives for refractory cases. 13 Further evidence comparing different treatment strategies for ES is needed to establish specific guidelines. It is worth noting that one-third of patients with chylous effusions secondary to SLE had concurrent hematological issues, with leukopenia being the most common.6,7,14-16 While a possible link exists between the simultaneous involvement of the hematological and lymphatic systems, further research is needed to clarify this association.
Another crucial aspect to consider when evaluating patients with systemic lupus erythematosus (SLE) who present with pancytopenia and fever is the possibility of macrophage activating syndrome (MAS). While the HLH-2004 criteria have traditionally been utilized to diagnose MAS (including symptoms such as fever, splenomegaly, cytopenia, elevated triglycerides/decreased fibrinogen, decreased NK cell function, increased ferritin, and elevated soluble IL-2 receptor levels, along with the demonstration of hemophagocytosis), their efficacy in identifying MAS secondary to autoimmune diseases has not been optimal. 17 As a result, alternative criteria have been proposed by EULAR-ACR and validated in patients with systemic juvenile idiopathic arthritis and pediatric SLE. These alternative criteria entail the presence of fever combined with high serum ferritin levels (>684 ng/ml) in addition to any 2 of the following criteria: platelet count <181 000, aspartate aminotransferase >48 U/l, triglycerides >156 mg/dl, or fibrinogen <360 mg/dl. The introduction of EULAR-ACR criteria has demonstrated enhanced sensitivity in identifying MAS among pediatric SLE and juvenile idiopathic arthritis cases. 18 However, there remains a requirement for specific criteria tailored to adult SLE and other rheumatological conditions. Despite our patient experiencing fever and elevated ferritin levels accompanied by a low platelet count, he did not meet the remaining criteria for the diagnosis of macrophage activating syndrome (MAS). Furthermore, his ferritin levels did not reach the diagnostic threshold for MAS, and the bone marrow biopsy did not reveal any evidence of hemophagocytosis.
In conclusion, we present the first case of massive right CPE as the initial presentation of overlap between SLE, APS, and ES. This is the first reported case of these pathological conditions occurring concomitantly. Our report underscores the challenges of diagnosing and managing severe lupus crisis patients, as multiple rare complications can coincide. The main learning points of this report are as follows: (1) Chylous pleural effusion can be a complication of SLE, and although it is not the most common etiology, it should be considered among the differentials. (2) Hematological compromise in SLE is common, and the sequential presentation of cytopenia should raise suspicion of ES. (3) Macrophage activating syndrome (MAS) is an entity that is often underdiagnosed in patients with autoimmune diseases. Despite the lack of specific criteria, it should be suspected in scenarios involving hyperferritinemia, fever, and cytopenias.
Identifying these complications as initial manifestations of SLE flares can aid in early diagnosis, timely treatment, and reducing sequelae incidence. Further studies with larger cohorts are necessary to comprehend the causative mechanism, intervening factors, diagnostic method, and best therapies in these problematic and rare clinical scenarios. Our report contributes to the limited sample size of patients with these conditions.
Footnotes
Acknowledgements
The authors acknowledge all the medical and nursing team that provided an excellent care for this patient.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author contribution
D.C.O was responsible for the conception of this article, patient care, case analysis, and manuscript writing. A.A.R. was responsible for case analysis and manuscript writing. C.R. and D.A. were responsible for patient care and manuscript revision.
Ethics approval and informed consent
Written informed consent was obtained from the patient for their anonymized information to be published in this article. Written informed consent is available upon request.