
Abstract
Background:
Juvenile dermatomyositis (JDM) is an autoimmune connective tissue disorder characterized by an inflammation of proximal muscles of both upper and lower limbs in children below the age of 18 years. The condition mainly involves the proximal muscles and skin but extra-muscular involvement such as the gastrointestinal tract, lungs, and heart are also common.
Case presentation:
We present a case of a 12-year-old south Asian male who developed weakness and muscular pain in all 4 extremities at 3 years of age. The condition gradually worsened recently, and the patient developed tender ulcerated skin nodules. Power in all 4 limbs was decreased and the patient was not able to perform his routine work such as combing of hair, closing a shirt button, and walking. Laboratory investigations revealed raised total leukocyte count (TLC) and erythrocyte sedimentation rate (ESR) and biopsy of the proximal muscles and skin lesions showed focal mild necrotic infiltrate involving nonnecrotic muscle fibers and calcinosis cutis respectively. A diagnosis of JDM was made and the patient was started on immunosuppressive therapy (steroids) and diltiazem.
Conclusion:
JDM shares clinical features with other autoimmune, genetic, and inflammatory conditions. Proper history, thorough clinical examination, and laboratory workup is needed to rule out other masquerading conditions. This case report also highlighted the importance of diltiazem in the treatment of calcinosis cutis which is more commonly seen in patients with dermatomyositis.
Introduction
Juvenile dermatomyositis (JDM) is a rare but chronic autoimmune and connective tissue disorder classified under the idiopathic inflammatory myopathies’ spectrum. Myositis-specific (MSAs) or associated autoantibodies (MAAs) have been linked to various clinical phenotypes of inflammatory myopathies and have been proven to help in the diagnosis of various subtypes. A study was conducted on 95 idiopathic inflammatory myopathies patients, MSAs and MAAs were measured in their sera. Dermatomyositis was the most prevalent subtype, 44% of the patient had MSAs while 23% had MAAs. 1 The clinical presentations of the disease are highly variable with prominent involvement of proximal muscles and skin. Extra muscular manifestations are also common and mainly involve the lungs, heart, and gastrointestinal tract. Early detection of extra muscular involvement and prompt management can decrease the mortality of the patients. JDM also has an increased risk of calcinosis cutis due to dystrophic calcification and multisystemic vasculitis but decreased risk of malignancy compared to adult dermatomyositis. 2 Even though it is rare, 85% of children with inflammatory myopathy have JDM. It is more common in females than males, with a 2.3:1 female-to-male ratio. Seven years is the average age for JDM symptom onset, and females may present with symptoms earlier. 3
The pathogenesis may involve genetic susceptibility as shown by HLA-DR3 having a strong connection with JDM. The proposed pathogenesis involves vascular injury due to autoantibodies against endothelial antigens. B and T cells as well as interleukins 6 and 17 are believed to play a potential role in JDM. 2 The diagnosis of the condition depends upon the clinical features supported by noninvasive investigations such as positive serology for autoantibodies, ultrasound, magnetic resonance imaging (MRI), X-ray, and nerve conduction studies. Diagnosis can be confirmed through muscles and cutaneous lesion biopsy which will show necrotic inflammatory changes in the proximal muscles and calcinosis cutis. Disease monitoring should be done with various tests such as pulmonary function tests, electrocardiograms, and swallowing studies to detect extra muscular involvement.
Treatment options for JDM include mycophenolate mofetil, hydroxychloroquine, azathioprine, tacrolimus, cyclophosphamide, intravenous immunoglobulin, infliximab, adalimumab, etanercept, abatacept, tocilizumab, steroids, and JAK kinase inhibitors. A few treatments have been studied with randomized control trials, including methotrexate, Ciclosporin, and Rituximab. 4 Recently diltiazem has been proven to be an effective treatment for calcinosis cutis associated with JDM. 5
The basic aim of reporting this case is to highlight the variety of JDM features that can occasionally make accurate diagnosis difficult, but thorough investigations aid in making the correct diagnosis. Additionally, this case report highlights the effectiveness of diltiazem in treating calcinosis cutis associated with JDM.
Case Report
A 12-year-old male child was brought to the outpatient department of a tertiary care hospital by his father due to progressive painful muscular weakness especially the proximal muscles of both upper and lower limbs and recurrent skin nodules. The patient developed muscular weakness at the age of 3 years which progressively worsened to the level that he is now unable to walk or perform routine work. The muscular weakness was associated with pain for which he used paracetamol and nonsteroidal inflammatory drugs (NSAIDs). He did not have a respiratory problem or difficulty swallowing food. The patient started developing skin nodules at the age of 8 years that are hard ulcerated and had a white chalky discharge. Initially, the skin nodules were episodic and now progressively worsened and involve both upper and lower limbs and axillae. The patient was unable to walk, comb his hair or change his clothes due to pain and weakness. The patient used painkillers on and off for body aches in the past and did not visit a hospital due to affordability issues. Family history was unremarkable for similar conditions.
On examination, the patient was vitally stable with a blood pressure of 100/70 mm Hg, pulse rate of 66 beats per minute, respiratory rate of 18 breaths per minute, and a temperature of 99°F. The patient was underweighting with a BMI of 16.5 kg/mm2. An erythema was visible covering the bilateral checks extending up to the auricular helix with nasolabial folds sparring (Figure 1).
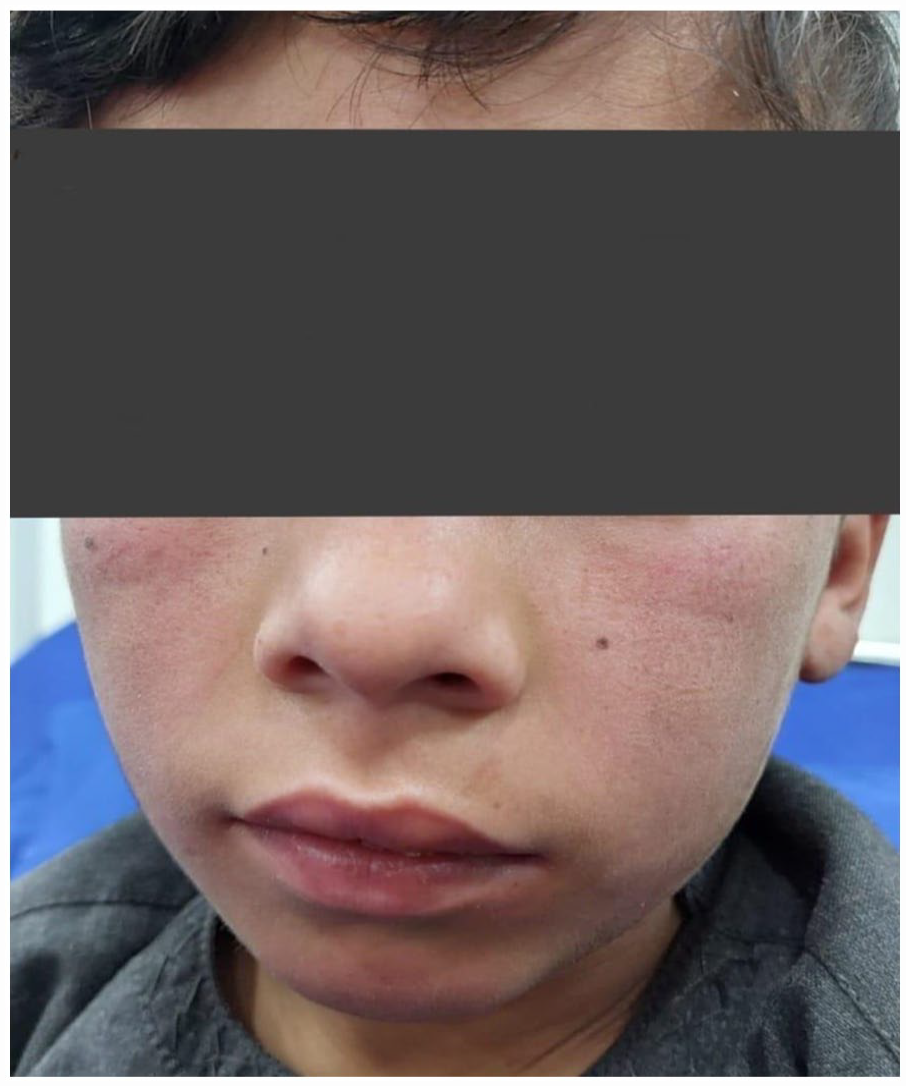
AN erythema of the face covering the bilateral cheeks with nasolabial folds sparring.
Multiple tender ulcerated skin nodules of about 1 × 1 to 2 × 2 cm with a white chalky discharge were visible over both upper and lower limbs including axillae (calcinosis cutis; Figure 2).

Ulcerated cutaneous nodules of 1 × 1 to 2 × 2 cm with chalky white discharge (Calcinosis Cutis) over the flexor surface of both upper limb and axillae (A), lateral aspect of the left knee (B), and bilateral legs (C).
The rest of the dermatological and nails examination was unremarkable. Musculoskeletal examination revealed tender muscles, and a power of 3/5 in all 4 extremities, and the patient exhibited a positive Gower’s sign (uses his hands and arms to stand up from a squatting position). The tone of both the upper and lower limb was decreased and the reflexes were normal. The patient’s cutaneous dermatomyositis disease area and severity index (CDAIS) score was calculated, the total activity score was 9 and the total damage score was 8. The patient was admitted to the dermatology unit for further workup, and initial laboratory evaluation revealed raised total leukocyte count (TLC), raised creatinine phosphokinase, and antinuclear antibody (ANA) was negative, while lactate dehydrogenase (LDH) level was elevated (Table 1).
Initial Laboratory evaluation.

Abbreviations: g/dL, gram per deciliter; IU/L, international units per liter; mcL, microliter; mEq/L, milli equivalent per liter; mg/dL, milligram per deciliter; U/L, units per liter.
Autoimmune workup was negative for anti-nucleosome antibodies, and Mi-2 except for the weakly positive lupus anticoagulant antibodies and positive anti-smooth muscle antibodies (ASMA). Chest radiograph and 2-dimensional echocardiogram were also normal. An X-ray of the lower limb was ordered which showed calcification in the soft tissues bilaterally (Figure 3).

X-ray of bilateral lower limb showing calcification in the soft tissues (calcinosis cutis) in both anterior (A) and posterior view (B).
A preliminary diagnosis of juvenile dermatomyositis was made and for confirmation, electromyography was done which showed a myopathy pattern. Magnetic resonance imaging (MRI) of the thigh was inconclusive Biopsy from the thigh muscles was taken which showed predominantly unremarkable skeletal muscle with a focal mild chronic inflammatory infiltrate involving nonnecrotic muscle fibers, skin biopsy showed subcutaneous deposits of calcium presenting calcinosis cutis, epidermis was unremarkable. The patient was started on tablet prednisolone 2 mg/kg once daily, IV methotrexate 10 mg/mA2 once weekly, and oral Diltiazem 2 mg/kg/day. The patient was asked for close follow-ups at 4, 8, and 16 weeks, he showed mild improvement in his muscular pain and weakness and was evaluated for drug side effects at each visit. On the 16th week of follow-up, the patient showed improvement in weakness the power of both shoulder and hip joint increased to 4/5, and the patient’s pain improved and was reported as 3/10. The patient was continued on diltiazem for calcinosis cutis and oral corticosteroid.
Discussion
Juvenile Dermatomyositis is a rare autoimmune disorder that primarily affects children and adolescents, with a higher prevalence in females. The precise etiology of this disorder is not clear; however, both immune dysfunction and environmental factors are thought to contribute to its etiopathogenesis. The clinical symptoms differ significantly patient-to-patient, but they generally involve muscle weakness and skin rashes. Weakness in the proximal muscles of the lower and upper extremities is commonly seen but not always the case, the lack of which has correlated with a milder manifestation of the disease. Extra muscular involvement of JDM includes the lungs, heart, and gastrointestinal system in the form of respiratory muscle weakness, interstitial lung diseases, myocarditis, and dysphagia. Fever, weight loss, fatigue, myalgias, arthritis, lymphadenopathy, and abdominal pain are constitutional symptoms known to be associated with JDM. 6
The diagnosis of JDM is made using the “Bohan and Peter criteria” developed in 1975. The diagnostic criteria include characteristic skin rash, symmetric muscle weakness of the upper and lower proximal muscles, increased levels of serum muscle enzymes, myopathic electromyography, and characteristic pathologic changes revealed by a muscle biopsy. Current practice reveals the necessity of broadening diagnostic criteria by incorporating new techniques, such us MRI and ultrasound, and the significance of skin disease in JDM. 7 The case presented above fulfilled all the components of the diagnostic criteria for JDM.
The differential diagnosis of JDM includes mitochondrial myopathies, infectious myopathies, autoimmune necrotizing myopathy, as well as Duchenne muscular dystrophy or Becker muscular dystrophy, systemic lupus erythematosus, and juvenile idiopathic arthritis. However, this patient’s family history, calcinosis cutis, negative ANA, other autoimmune antibodies, and biopsy findings ruled out other similar conditions. Interestingly our patient presented with some features of JDM, that is, the gradual increase of muscular weakness, the calcinosis cutis, and the patient history supported the diagnosis of JDM.
However, the defining characteristics of JDM that differentiate it from other Idiopathic Inflammatory Myopathies are the presence of distinctive skin rashes such as Heliotrope Rash and Gottron’s signs, as well as other dermatological features like erythema of extensor surfaces of joints, and calcifications on the skin and subcutaneous tissues. 8 Interestingly, our patient presented with only some of the symptom characteristics of JDM. He suffered from muscle aches and weakness since age 3, which gradually worsened to the point of hindering his ability to perform ADLs. Although the physical examination revealed a bilateral rash on both cheeks, he did not have Gottron’s papules. He also had clinical features consisting of calcinosis cutis in both upper and lower limbs along with an elevated level of serum muscle enzymes, characteristic biopsy findings, and, electromyography showing a myopathy pattern. This case of JDM, therefore, underscores the importance of realizing the heterogeneity of symptoms that can manifest in this autoimmune disease.
There are a number of cases reported earlier showing similar clinical features and different associations manifesting JDM, the following also shows that positive serology is not needed for the correct diagnosis and the confirmation must be done by muscles or skin biopsy (Table 2).
Shows typical and associated clinical features and treatment options of JDM.

Treatment for Juvenile Dermatomyositis has predominantly relied on corticosteroids. Other non-steroidal treatment approaches have included Methotrexate, Cyclosporin, Cyclophosphamide, and intravenous immunoglobulin. For patients who specifically suffered from calcinosis cutis as a complication of JDM, diltiazem may be an effective form of treatment. Diltiazem acts as a calcium channel blocker to reduce the deposit of calcium in soft tissues. Overall, the prognosis of JDM is variable based on the patient’s specific symptoms and the time to treatment. Nonetheless, the advent of corticosteroid therapies has helped reduce the mortality rate to 10%. 13
Conclusion
This case emphasizes the significance of recognizing the variety of symptoms that might appear in conditions like JDM, which can mimic other similar conditions as in our patient. To lessen the burden of long-term complications, it is crucial to diagnose the condition early on in the course of the disease with a proper history, thorough clinical examination, and laboratory workup. Additionally, long-term treatment with diltiazem is effective in calcinosis cutis associated with dermatomyositis without any adverse effects.
Footnotes
Author Contributions
Q.A.K and T.K collected the data, C.F, F.A.H, H.P, H.S, M.A, P.A wrote the original manuscript. Q.A.K did the critical review and final editing of the manuscript. All authors reviewed and approved the final version of the manuscript before submission.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.