
Abstract
Background:
Neurofibromatoses are a rare group of autosomal dominant tumor suppressor phacomatoses syndromes. Neurofibromatosis type 1 (NF1 or Von Recklinghausen’s disease) is the most commonly found type of neurofibromatosis, and constitutes the most commonly found autosomal dominant disease of the nervous system.
Case presentation:
We report a case of a 14-year-old boy who reported a 3-year-history of a slowly enlarging right lateral cervical mass. He has a medical history of a progressive limping gait disorder with scoliotic attitude. MRI identified a dumb-bell shaped intradural right cervical process through right paravertebral gutter on C2 to C4, a second intradural dorsal mass with the same characteristics through left paravertebral gutter on D4 and D5 and a large tissue-like mass infiltrating the lumbosacral subcutaneous soft tissues. A Surgical excision of the cervical and lumbar masses was performed with a good outcome after surgical excision.
Conclusions:
This case illustrates the need for a collaboration of both neurological and head and neck surgeons in terms of managing difficulties related to a cervical neurofibroma. Benign plexiform neurofibromas are rapidly growing tumors, particularly in children and adolescents, which makes all the importance of early detection and appropriate treatment. Repeated interventions are usually needed in order to adapt and stabilize the tumors extension.
Background
Neurofibromatoses, as they involve structures arising from the embryonic ectoderm, are a rare group of autosomal dominant tumor suppressor phacomatoses syndromes. This heterogenous hereditary group of disease share as part of their clinical presentations peripheral nerve sheath tumors. Neurofibromatosis type 1 (NF1 or Von Recklinghausen’s disease) is the most common type of neurofibromatosis, and autosomal dominant disease of the nervous system.1,2
We herein report a case of a 14-year-old patient with concomitant cervical and dorsal spinal neurofibromas revealed by a severe motor deficit, with a good evolution after surgical excision.
Case Presentation
We report a case of a prepubertal 14-year-old patient who was admitted to our hospital with a 3-year-history of a slowly enlarging right lateral cervical mass. The major symptom was a progressive installation of a limping gait disorder with scoliotic attitude.
Past medical history found a lumbosacral hypertrichosis onset at birth, treated malaria, grass, and sand allergy. The brother was followed for homozygous sickle cell disease. There are no cases of familial neurofibromatosis.
On admission, vital hemodynamic and respiratory signs were within normal range. Physical examination showed a painful, soft and mobile cervical mass which measured 6 × 6 cm and was tender upon palpation. There was not no cervical adenopathy. Neurological examination found a bilateral motor impairment: motor power was grade 2 on the Medical Research Council (MRC) scale bilaterally. A tetra-pyramidal syndrome with spasticity was found with exaggerated right deep tendon reflexes. No sensitive nor sensorial deficit was found. Café-au-lait spots were observed in the body.
Spinal Magnetic Resonance Imaging (MRI) identified a dumb-bell shaped and well limited intradural right cervical process, hypo-signal in T1 sequence and intermediate signal in T2 sequence. Following administration of gadolinium, the lesion showed marked enhancement. The mass expands through right paravertebral gutter on C2 to C4 and measured 64 × 46 × 65 mm (Figure 1).

T1 MRI sequence showing a hyposignal well limited dumb-bell shaped intradural right cervical process.
A second intradural dorsal mass with the same characteristics was visualized through left paravertebral gutter on D4 and D5 measuring 42 × 26 mm (Figure 2).

T2 MRI sequence showing an intradural dorsal mass in slight hyper-signal visualized through left paravertebral gutter on D4 and D5.
On the lumbar level, we found a large tissue-like mass infiltrating the lumbosacral subcutaneous soft tissues which showed hypo-signal in T1 sequence and a slightly hyper-signal in T2 STIR sequence. The mass arrived in depth contact with the superficial aponeurosis of the paravertebral muscles and measured 124 × 48 × 160 mm (Figure 3). It is associated with a second skin lesion measuring 8 × 28 mm and which have the same characteristics.

T2 STIR MRI sequence showing a large hyper-signal tissue-like mass infiltrating the lumbosacral subcutaneous soft tissues.
In consideration of the several symptoms and the imaging findings, surgical excision of cervical and lumbar masses was decided by both neurosurgical and the otolaryngology head and neck surgery team. For perioperative blood saving concern, it was decided to schedule the various surgical procedures in a deferred manner in order to limit blood loss.
Priority was given to neurosurgical excision via the posterior approach of the compressive neck mass to relieve compression and improve motor function immediately. Through an incision from the occiput to C5, we performed a dissection ablation of a hard, well-defined yellowish encapsulated tumor emerging from C2-C3 in the right paravertebral gutter. This tumor continued through the C2-C3 intra spinal conjugation hole. A C2-C3 laminectomy is performed, then dissection of the tumor and ablation of this one by morcellation.
In a second step, 3 days later, the rest of the cervical mass was approached anteriorly by the otolaryngology and head and neck surgery team. Through a right Paul-Andrey incision, we dissected along the deep surface of the sternocleidomastoid muscle until the discovery of a bulky whitish, hard, encapsulated mass pushing back the right jugulo-carotid axis, spinal nerve and digastric muscle. A subtotal excision of the mass was performed (Figure 4).
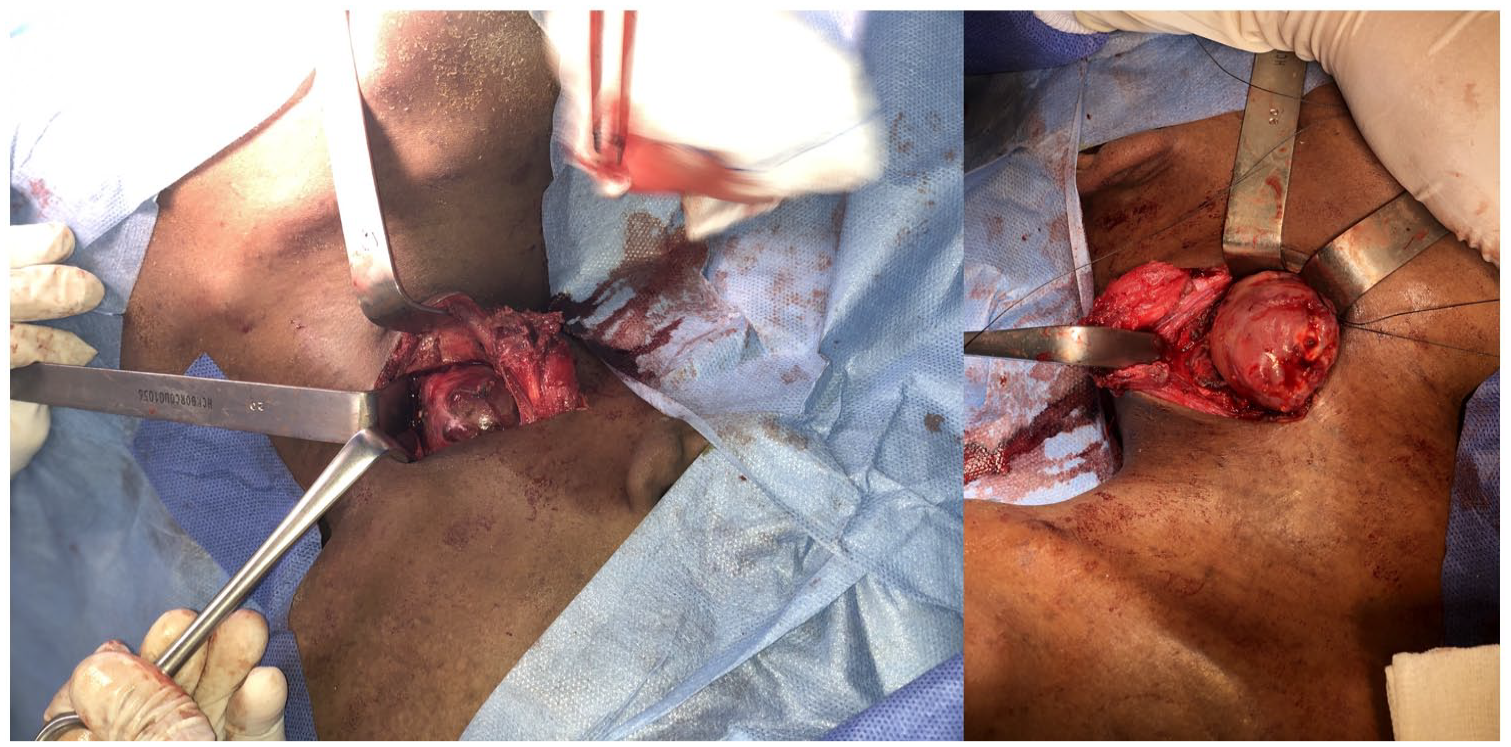
A subtotal cervical bulky whitish, hard, encapsulated mass excision approached anteriorly by the otolaryngology and head and neck surgery team through a right Paul-Andrey incision.
Postoperative outcome was good and, the patient progressively recovered from his motor deficit, which was evaluated at left 4/5 (right = normal) MRC on day 5 post op, then 5/5. He was discharged from the hospital in the fifth day; Concertation with oncologists, radiotherapy or chemotherapy was not needed.
Pathology with immunocytochemical study describes an aspect of a poorly limited tumoral proliferation, made up of slender and flexuous fusiform cells, grouped in bundles. The nuclei are fusiform or oval of uniform size with regular chromatin. These tumor cells express PS100 diffusely. The fibroblast component cells express CD34. An absence of Acute myeloid leukemia (AML) expression is noted. KI67 is estimated at 5%. This aspect is in favor of a neurofibroma.
In a third step, 18 months later, we proceeded to the dissection ablation of a lumbar tumoral mass (relating to the dorsal neurofibroma) emerging from L4-L5 of firm consistency, whitish color, anchoring itself on a nerve root of approximately 2 cm. The pathology study found a morphological aspect of a plexiform and diffuse neurofibroma of the lumbar seat.
Postoperative outcome was good, no sensitive nor motor deficit was declared. The patient recovered well, and was discharged from the hospital 6 days after the intervention.
Discussion
Neurofibromatoses are a rare group of autosomal dominant tumor suppressor syndromes. This heterogenous hereditary and yet well-defined group of phacomatoses share as part of their clinical presentations peripheral nerve sheath tumors. NF1 (or Von Recklinghausen’s disease) is the most commonly found type of neurofibromatosis, and constitutes the most common autosomal dominant disease of the nervous system with a prevalence that ranges from 1 to 2 in 4000.1,2 Known for their predilection for nervous system involvement, Neurofibromatoses, particularly NF1, include a wide range of systemic non-neurological symptoms. 3 The causative gene for NF1 was mapped to human chromosome 17 and identified as the NF1 gene. This gene encodes for neurofibromin, a tumor suppressor protein that has various functions including participation in several signaling pathways. This protein is predominantly expressed in neurons, Schwann cells, oligodendrocytes, astrocytes and leukocytes, which explains the systemic nature of this disease and the predominance of neurological manifestations.3,4
The clinical diagnosis criteria for NF1 were based in the modern nomenclature and history from the 1987 Bethesda National institute of Health consensus development conference on Neurofibromatosis, which are highly specific and sensitive in most patients. 5 In 2021 a multistep process and multispecialty effort involving Neurofibromatosis (NF) experts, non-NF experts, patients and patient advocacy groups, an international consensus recommendation was published proposing A revised diagnostic criteria (Table 1) for neurofibromatosis type 1. 6
Revised diagnostic criteria for neurofibromatosis type 1 (NF1).

If only café-au-lait macules and freckling are present, the diagnosis is most likely NF1 but exceptionally the person might have another diagnosis such as Legius syndrome. At least one of the 2 pigmentary findings (café-au-lait macules or freckling) should be bilateral.
Sphenoid win dysplasia is not a separate criterion in case of an ipsilateral orbital plexiform neurofibroma.
The diagnosis of NF1 was made in our patient who met 3 of the revised diagnostic NF1 Criteria, as the following:
Multiple Café-au-lait macules (>50) all over the body, from which at least 10 were over 5 mm in greatest diameter.
Multiple Lisch nodules (>10) identified by slit lamp examination
Multiple neurofibromas, including a cervical plexiform neurofibroma
Spinal neurofibromas that arises from the spinal nerve root sheath are a the most common localization in patients with NF1, and occur in as high as 80% of patients during their lives.7,8 Multiple spine regions localization is common, and up to 56% of patients between 10 and 18 years of age, as in the case of our patient, had 2 or more spinal localizations. 8 Cervical spine involvement is usually asymptomatic, but can progress with time into structural abnormalities such as widening of the neural foramens, vertebral scalloping, osteolysis, kyphotic deformity and severe neurological symptoms in case of spinal cord compression like in our case.9,10
Plexiform neurofibromas are an invasive intra neural tumors with massive growth, and an increased risk of malignant transformation. Those tumors have greater rate of mortality and morbidity. 11
The surgical management of Von Recklinghausen’s disease remains a challenge in different aspects of the disease. In the literature, we found that approximately 26% to 50% of patients carry skeletal deformities, which increase the risk of developing neurologic sequelae.12,13 One other option proposed to inoperable plexiform neurofibromas is using a selective inhibitor of mitogen-activated protein kinase kinase (Selumetinib), therefore inhibiting the RAS (Rat Sarcoma virus) pathway. 11
Fortunately, the chance of postoperative regrowth is very low, therefore perhaps explaining why surgeons seem to maintain the surgical excision as the treatment of choice for Plexiform Neurofibromas.14 -18
Conclusion
This case illustrates the need for a strong collaboration between neurosurgical and head and neck surgeon teams in terms of managing difficulties related to a cervical neurofibroma. Benign plexiform neurofibromas are rapidly growing tumors, particularly in children and adolescents, which makes all the importance of early detection and appropriate treatment. Repeated interventions are usually needed in order to adapt and stabilize the tumors extension.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Ilias Tahiri, Abderrahim Bourial, Wahib Lahlou and took part in taking care of the patient, researching the bibliography and writing the article. Amal Hajjij took part in the design, editing the article and approved the final manuscript. Abderrahmane Al Bouzidi contributed to the pathological findings of this case and approved the final manuscript. Mounir Rghioui, Mohamed Zalagh, Abdessamad El Azhari, Fouad Benaariba approved and participated in the process of validating the final manuscript.
Availability of Data and Material
On demand, by asking the corresponding author.
Ethical Approval
Not applicable.
Consent to Participate
Orally.
Consent for Publication
Orally.
Code Availability
Not applicable.