
Abstract
Systemic lupus erythematosus (SLE) involves dysregulation of the immune system, consequently affecting multiple organ systems, including the cardiovascular, neuropsychiatric, renal, and musculoskeletal systems. Optic neuritis and intracranial hypertension are conditions that rarely occur in SLE, and their coexistence has not been reported to date. Herein, we report the first case of a patient who was diagnosed with SLE complicated by concurrent intracranial hypertension and bilateral optic neuritis. An 11-year-old Thai girl had a low-grade fever, discoid rash, oral ulcer, chronic headache, and fluctuating diplopia. She experienced bilateral vision loss just before presentation. She was diagnosed with juvenile SLE. We believe that her headache, which was probably a symptom of optic disc edema, was due to intracranial hypertension. Furthermore, she exhibited vision loss and color vision deficit and was diagnosed with bilateral optic neuritis. Her condition improved on treatment with corticosteroids (intravenous pulse methylprednisolone for 3 days, followed by 1 mg/kg/day oral prednisolone tapered over 3 months). The occurrence of optic neuritis and intracranial hypertension during an active SLE inflammation and a rapid response to high-dose corticosteroids support the fact that SLE was the etiology of these neuropsychiatric conditions. Early diagnosis and prompt treatment in such cases can lead to favorable outcomes.
Background
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that affects multiple organ systems and has a relapsing and remitting course. The prevalence of SLE varies across literature, ranging from 72.8 to 178 cases per 100 000 patients yearly. 1 It commonly occurs in patients of African and Asian descent. 2 The median age of onset is late teen to early 40s, and women are 9 times more susceptible than men. Major histocompatibility complex genes, such as human leukocyte antigen (HLA)−A1, B8, and DR, are associated with disease occurrence. 3 In SLE, complex dysregulation of the immune system affects multiple systems of the human body. Various autoantibodies, such as anti-double-stranded DNA, anti-Ro, La, Sm, nucleosome, N-methyl-D-aspartate receptor, and phospholipid, have been identified to cause the disease. 4 The pathogenesis involves immune complex deposits in multiple organs, stimulating complements, and other mediators of inflammation. 5
The presentation and severity of SLE vary across individuals. Neuropsychiatric SLE (NPSLE) is a common presentation of SLE; however, optic neuritis and intracranial hypertension have rarely been reported to coexist with it. In SLE, the prevalence of optic neuritis is 0.6% to 1% 6 and that of intracranial hypertension is 1% to 1.5%.7,8 To the best of our knowledge, there are no documented case reports of the coexistence of both conditions in SLE.
Herein, we describe the first case of a patient with a new onset of active SLE and concurrent occurrence of optic neuritis and idiopathic intracranial hypertension, aiming to present the clinical presentation, physical examination, investigations, treatment, and subsequent outcome of this rare case.
Case Presentation
A previously healthy 11-year-old Thai girl had a low-grade fever for 1 month. Ten days prior to admission to our hospital, she had a high-grade fever, malaise, and loss of appetite. Subsequently, she developed a throbbing headache with fluctuating double vision. Finally, she lost her eyesight 1 day before admission. On admission, her body temperature was 38.3°C, and other vital signs were unremarkable. Physical examination revealed that she had no malar rash, alopecia, or arthritis. However, she had a discoid rash (hyperpigmented plaque with follicular plugging) on her left ear, painless ulcer on the hard palate, hepatosplenomegaly (14 cm), proximal muscle weakness of grade 4 in the upper extremities, and hyporeflexia in the upper extremities with areflexia in both the lower extremities.
Eye examination revealed that her vision in the right eye was graded as hand motion with good projection of light and that in the left eye as finger counting at 2 feet. Her ocular alignment showed esotropia of 30 prism diopter using the Hirschberg technique. Limited abduction was more severe in her left eye than in her right eye (Figure 1). Bilateral abducens nerve palsy caused limited abduction in both eyes. The anterior segment showed unremarkable findings, except for a relative afferent pupillary defect of grade 2 in the right eye. Eyeground examination showed severe bilateral disc edema, peripapillary hemorrhage, cotton wool spots, and subretinal fluid in the macular area of the left eye (Figure 2).

Gazes positions showing bilateral limited abduction in the left and right photos.

Optic disc photo of both the eyes showing optic disc edema with peripapillary hemorrhage and cotton wool spots. Macular edema is noted on the left eyeground examination.
Laboratory test results on day 2 of admission showed the following findings: pancytopenia (hemoglobin level: 7.9 g/dL, hematocrit level: 23 g/dL, white blood cell [WBC] count: 2100/µL, platelet count: 81 000/µL); direct Coombs test, 3+; anti-double-stranded DNA positivity, 1:80; antinuclear antibody >1:5; 120 homogeneous pattern; low complement C3 level, 0.13 g/L; low complement C4 level, 0.06 g/L; proteinuria with hematuria; negative anti-cardiolipin IgG finding; and positive anti-beta 2 glycoprotein 1 IgG/IgM/IgA finding. According to the recent American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) 2019 criteria, 9 the diagnosis was juvenile SLE in this case.
Her headache may have been symptomatic of an underlying neuropsychiatric complication of intracranial hypertension, with increased intracranial pressure, consequently leading to bilateral optic disc edema. Bilateral abducens nerve palsy caused limited abduction in both eyes. More investigations performed on day 2 to day 4 of admission revealed the following results. Magnetic resonance imaging (MRI) revealed no mass-like lesion, but signs indicative of an increased intracranial pressure were detected (Figure 3). Magnetic resonance venography showed no venous sinus thrombosis or stenosis, and magnetic resonance angiography findings were unremarkable. Lumbar puncture showed an opening pressure of 60 cmH2O and a closing pressure of 19 cmH2O. Cerebrospinal fluid (CSF) analysis revealed a WBC count of 2 cells/µL, a red blood cell count of 1 cells/µL, and glucose concentration of 58 mg/100 mL (dextrostix: 117 mg%), which was unremarkable. The patient tested negative for oligoclonal band, autoimmune encephalitis profile, and other infections. Finally, on day 4 of admission, she was diagnosed with intracranial hypertension according to the modified Dandy criteria. 10

Magnetic resonance imaging of the brain and orbit with gadolinium contrast showing faint enhancement of both optic nerves (top photo showing axial view, and the bottom photo showing coronal view).
Concurrently, bilateral vision loss was noted. The differential diagnoses were bilateral macular ischemia, bilateral optic neuritis, and posterior visual pathway disorder, such as occipital lobe ischemia. She had color vision deficits in both eyes, as determined by the Ishihara test. The central visual field (CTVF 24-2) could not be interpreted owing to fixation loss in both eyes. Optical coherence tomography angiography of the macula revealed a normal foveal avascular zone (Figure 4). Fluorescein angiography revealed late capillary leakage, peripapillary staining, no capillary drop-out area, and normal foveal avascular zone. MRI of the brain and orbit with gadolinium contrast showed faint enhancement and enlargement of both the optic nerves. No hypersignal T2 was detected along both the optic nerves. Brain parenchyma was normal without any non-specific white matter changes (Figure 3). Visual evoked potential (VEP) showed a significantly delayed P2 without decreased amplitude in the right optic nerve compared with that in the left optic nerve (Figure 5). In this case, the diagnosis was optic neuritis of both eyes.
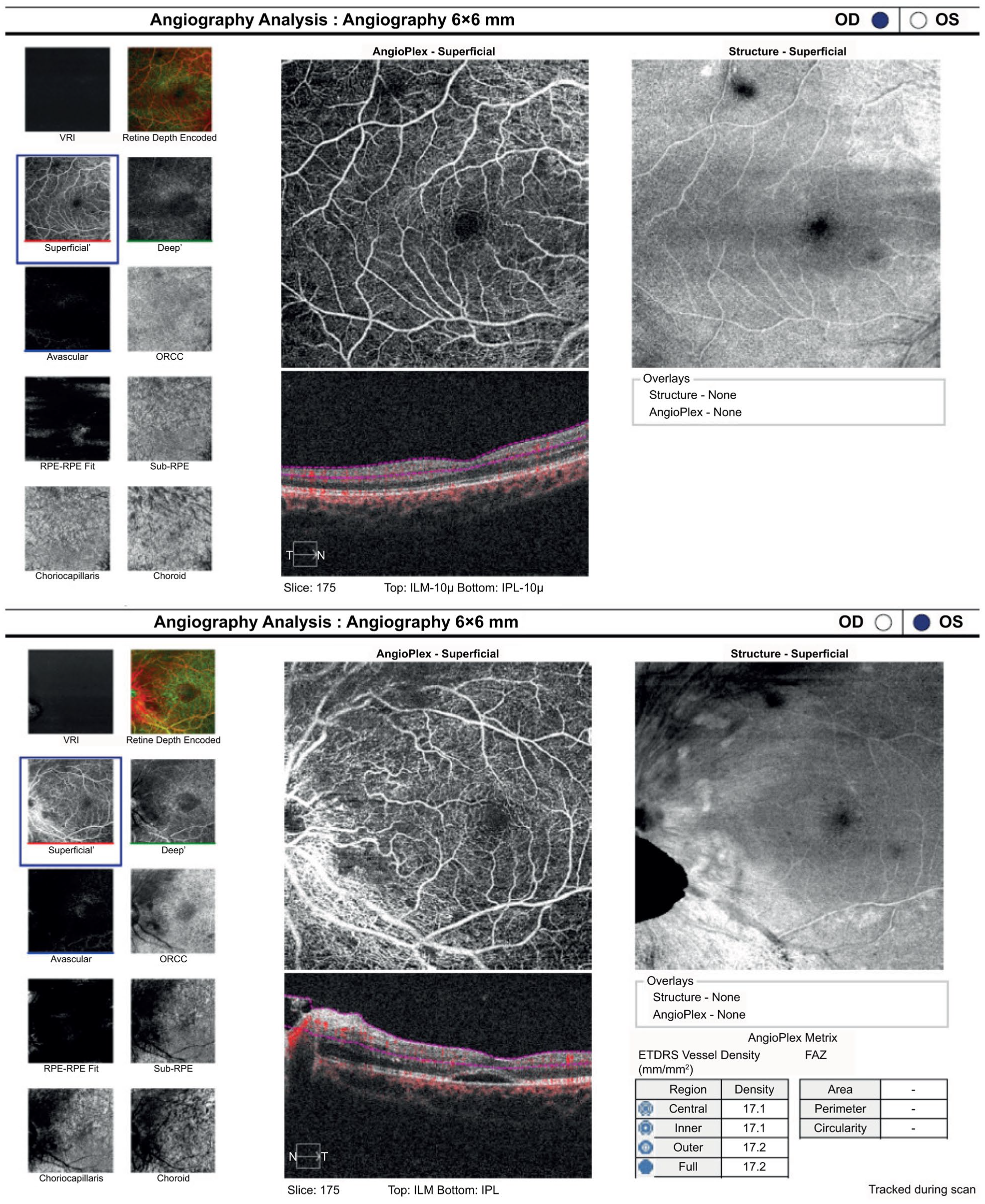
Optical coherence tomography angiography of both the eyes showing a normal foveal avascular zone (top photo representing the right eye and bottom photo representing the left eye). A hyporeflective space underneath the submacular area of the left eye is noted.

Flash visual evoked potential (VEP) showing delayed P2 in both eyes with more prominence on the right. Top photo showing the VEP graph and bottom photo showing its results.
Persistent proteinuria >0.5 g/day and hemoglobinuria suggested a diagnosis of lupus nephritis according to ACR criteria. After 1 month of admission, a renal biopsy was performed. The result revealed focal lupus nephritis, class III(A).
The standard treatment of intracranial hypertension in patients with SLE is high-dose corticosteroids. Similarly, steroids can be administered for treatment of SLE with optic neuritis. Therefore, intravenous pulse methylprednisolone 30 mg/kg/day was administered for 3 consecutive days to treat both these conditions, and treatment was continued with 1 mg/kg/day of oral prednisolone, whose dosage was tapered over 3 months. For lupus nephritis class III(A), we initiated an induction treatment with intravenous cyclophosphamide at 600 mg (500 mg/m2/dose) monthly for 6 doses and administered mycophenolate mofetil along with corticosteroid dosage tapering over the treatment course for maintenance.
Her eye condition gradually recovered. One week after treatment, which was 2 weeks from her initial visit to our hospital, papilledema improved. Her best-corrected vision was 20/400 in the right eye and 20/30 in the left eye. Her right eye regained full ocular abduction, and her left eye had 20% limitation of abduction. A drastic response after treatment with steroids supported the diagnosis of optic neuritis. After a 1-month follow-up, her best-corrected vision recovered to 20/70 in the right eye and 20/40 in the left eye, and eye movement recovered completely. At 2 months, her corrected vision was 20/50 in her right eye and 20/30 in her left eye. Five months later, she developed faint posterior subcapsular cataracts in both eyes due to prolonged corticosteroid treatment, with best-corrected vision of 20/50 in both eyes.
Discussion
We report a case with new onset of active SLE complicated by optic neuritis and intracranial hypertension. These complications rarely occur in SLE, and this is the first case of concurrent presentation. After diagnosis, prompt treatment with high-dose corticosteroids lead to clinical improvement. A problem-based approach in this case was the key to determining the diagnosis.
The severity of SLE depends on the organs that are involved in the immune processes. SLE may be difficult to diagnose because its early signs and symptoms are not specific and can present similar to those of other diseases. Nearly one-third of the patients with SLE have ophthalmic involvement. 11 Its most common association is keratoconjunctivitis sicca or dry eye syndrome, whereas the most severe ocular complication is retinal or optic nerve involvement. 12
The neuropsychiatric symptoms in NPSLE are a frequent cause of morbidity in these patients. According to the findings reported in the literature, the prevalence of NPSLE varies from 16% to 75%.13,14 Despite rapid diagnosis and treatment, morbidity continues to remain high.15,16 In 1999, the American College of Rheumatology Research Committee classified NPSLE on the basis of central and peripheral involvement. 17 Idiopathic intracranial hypertension, which is a cause of headache, and optic neuritis, which is a presentation of demyelinating disorders, are associated with NPSLE with central involvement.
Headache is a common presentation and is frequently found in approximately 38.3% of juvenile patients with SLE and 72.5% of patients with NPSLE. 18 Intracranial hypertension is an uncommon cause of intractable headache, presenting as the initial symptom of active SLE. Many studies have reported intracranial hypertension as the first presentation of juvenile-onset SLE.19 -24 Idiopathic intracranial hypertension was first reported by Bettman et al in 1968. 25 Since then, several SLE cases have been reported to be associated with intracranial hypertension. However, to date, the mechanism of increased intracranial pressure remains unknown but is hypothesized to be associated with the vasculitis process, immune complex deposits, or direct antibody injury to the arachnoid villi, which block CSF absorption and eventually cause elevated CSF pressure. In our case, the patient’s headache may have been a symptom of an underlying neuropsychiatric complication of intracranial hypertension, with increased intracranial pressure, consequently leading to bilateral optic disc edema.
As there are no specific criteria for the diagnosis of intracranial hypertension in patients with SLE, we adopted the modified Dandy criteria. 10 The following criteria were fulfilled in our case: symptoms and signs of increased intracranial pressure, absence of neurologic signs, CSF pressure >25 cmH2O, normal CSF profile, and normal neuroimaging. Further, MRI of the brain with magnetic resonance venography can support the diagnosis. Intracranial mass-like lesion and venous sinus thrombosis or stenosis must be excluded. The most serious complication is visual loss from severe papilledema called fulminant intracranial hypertension. 26 As the pathophysiology of high intracranial pressure involves excessive production of inflammatory cytokines and clogging of the arachnoid villi by inflammatory cells, the mainstay of treatment is high-dose corticosteroids, which can drastically resolve symptoms. It can be difficult to confirm that SLE was the cause of intracranial hypertension because currently there is no biologic marker for this condition. Furthermore, we lacked pathologic evidence. The clinical clue that supports the hypothesis that intracranial hypertension was due to SLE is that clinical onset was concurrent with the patient’s active SLE condition and the rapid clinical response to intravenous corticosteroid treatment.
Bilateral visual loss in patients with SLE can be due to various causes. The differential diagnoses in such cases could be bilateral optic neuropathy, bilateral retinal involvement, or posterior visual pathway disorder. The most likely etiology of visual loss is bilateral optic neuritis. The prevalence of SLE with optic neuritis is quite low at 0.6% to 1%. 6 The presenting visual acuity is usually worse than 20/200 depending on the degree of axonal loss. Optic neuritis can present either with unilateral or bilateral involvement, and it can occur as the initial presentation of active SLE or along the course of the disease. It is predominantly prevalent in women than in men. Variable responses of visual outcome after the treatment may range from finger counting to full recovery. Prognosis is not quite good as that in inflammatory demyelinating optic neuritis because it may be affected by the ischemic process; in particular, occlusive vasculitis affects small arterioles that cause secondary demyelination and axonal loss. In our case, evidence from MRI and VEP findings supports the diagnosis of optic neuritis. More than 90% of optic neuritis cases show optic nerve enhancement in T1 with the fat suppression technique. 27 A hyper-T2-weighted signal can be observed and be persistent even after long-term follow-up. The latency of the VEP accurately reflects the amount of demyelination, whereas the amplitude correlates with axonal damage in optic neuritis. Visual outcomes depend on the degree of axonal loss. 28 Early diagnosis and treatment with high-dose corticosteroids are associated with good visual outcomes. Benefits from other immunosuppressive agents, such as cyclophosphamide, cyclosporine, methotrexate, and azathioprine, have also been reported.29,30 Neuromyelitis optica spectrum disorder-associated optic neuritis should be excluded because only a few case reports have reported neuromyelitis optic antibodies in SLE cases.31 -33 It is related to poor visual outcomes and requires long-term immunosuppressive treatment to control relapse. In our case, the patient most likely had SLE-associated optic neuritis rather than idiopathic optic neuritis because the condition occurred during an active SLE inflammation, with poor visual acuity at onset that gradually improved after high-dose steroid treatment.
Macular ischemia can also be an etiology of acute visual loss in SLE. 34 The pathophysiology involves the absence of perfusion in the macular area due to the effects of vasculitis. It is associated with manifestations of severe retinal vasculitis; however, rapid improvement with steroid treatment made this differential diagnosis less likely in our case.
Intracranial hypertension can explain the characteristics of visual loss. An acute visual loss is less common than a chronic loss in cases of increased intracranial pressure. Two mechanisms of vision loss have been proposed: outer retinal change and optic neuropathy, which can occur alone or concurrently. The presence of subretinal fluid, chorioretinal fold, and peripapillary chorioretinal fold, which can lead to disruption of the photoreceptor layers, is the likely cause. 35 The outer retinal causes are usually reversible, typically 1 to 4 weeks after treatment. The pathophysiology of acute visual loss in intracranial hypertension, called fulminant intracranial hypertension, resulting from optic neuropathy is not well-understood and can be barely detected because of the obscure detection of disc edema-related structural changes. Retinal thinning reflects both optic nerve damage and optic disc edema improvement. The thinning of the macular ganglion cell layer-inner plexiform layers has a strong relationship with visual loss in optic nerve diseases. The short duration of recovery has made this etiology less likely. 35
Posterior visual pathway abnormality could also be a cause of bilateral visual loss. In the absence of other neurological signs, the lesion is usually bilateral occipital lobe stroke in such cases. However, nearly 3% to 20% of patient with SLE too experience stroke events, especially in the first 5 years of diagnosis. The potential mechanisms of cerebrovascular disease in SLE include hypercoagulable states, cardiogenic embolism, premature or accelerated atherosclerosis, and rarely vasculitis. 36 However, rapid vision response after steroid treatment and negative neuroimaging findings in the posterior visual pathway made this etiology less likely.
The outcomes in SLE complicated by increased intracranial pressure and optic neuritis may be good with early steroid therapy as in our case. The complication of delayed or no treatment in such cases was degradation of visual function. Furthermore, this is the first case of its kind; the long term outcome after successful treatment can be described only after further follow-up.
Our approach in this case has some limitations. There is no definite evidence that SLE was the etiology of optic neuritis and intracranial hypertension in our case. Examination of a pathologic specimen may be crucial for supporting this hypothesis. Future studies on new biomarkers are warranted to predict the occurrence of complications in patients with SLE. Moreover, discovery of new medications could improve disease control and lead to better outcomes.
Conclusions
This is the first case report of a patient with SLE who had concurrent optic neuritis and intracranial hypertension. Although rare, both conditions can occur concurrently, as noted in our case. Contrastingly, either of the conditions can occur alone in early- to late-onset SLE presentations. A problem-based approach in complicated cases leads to assessment of underlying etiologies, thorough investigations, and appropriate management of the condition. Several evidences support early treatment for better outcomes. Future studies can contribute new findings to determine the exact pathophysiology and specific treatment.
Learning Points
Intracranial hypertension and optic neuritis, although rare, can occur concurrently in patients with neuropsychiatric systemic lupus erythematosus.
Footnotes
Acknowledgements
We thank Ratanasuda Thongruay, MD, an ophthalmology resident of the Phramongkutklao Hospital, for assistance in photo preparation and sequencing.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Worapot Srimanan, MD, treated the subject and collected the clinical data. Worapot Srimanan, MD, wrote the manuscript, and Somboon Panyakorn, MD, revised the manuscript. All authors approved the final version of the manuscript. The authors agree to be responsible for all aspects of this work.
Study Approval Statement
This study was conducted following the standards issued by the World Medical Association’s Declaration of Helsinki guidance. This protocol was reviewed and approved by the Institutional Review Board of the Royal Thai Army Medical Department (approval number: S095h/64_Exp).
Patient Consent
Written informed consent was obtained from the patient’s legal guardian for the publication of this case report and any accompanying images.