
Abstract
Introduction:
For many years, congenital panhypopituitarism has been recognized to cause infantile cholestasis. However, the isolated cortisol deficiency as a cause of cholestasis and liver failure was rarely reported.
Case description:
A 32-days old male infant presented to the hepatology clinic with infantile cholestasis. His initial workup revealed alanine transaminase (ALT) level of 138 U/L, aspartate transaminase level of 76 U/L, total bilirubin (T.Bil) of 103 mmol/L, direct bilirubin of (D.Bil) 83 mmol/L, gamma-glutamyl transpeptidase (GGT) level of 28 U/L with normal prothrombin time (PT) of 13 seconds. One week later, the patient developed severe bronchiolitis necessitating mechanical ventilation associated with acute liver failure and worsening cholestasis. His ALT increased to 303.5 U/L and direct bilirubin increased to 204 mmol/L with prolongation of PT to 18.9 seconds reflecting derangement in synthetic liver functions. There was associated hypoglycemia, hyponatremia and high normal potassium level with a picture of adrenal insufficiency. Hormonal workup and genetic testing revealed isolated cortisol deficiency with a novel homozygous mutation c.763_764delAT (p. Met255ValfsX17) in Melanocortin 2 receptor gene (MC2R) and the patient was diagnosed as familial primary glucocorticoid deficiency. The patient was maintained on cortisol replacement therapy with the resolution of cholestasis and normalization of liver functions.
Conclusions:
Patients presenting with infantile cholestasis associated with documented hypoglycemia should alert pediatricians about the possibility of familial glucocorticoid deficiency and prompt investigation of adrenal function should be considered. Cortisol replacement therapy leads to the resolution of cholestasis.
Introduction
Familial glucocorticoid deficiency (FGD; MIM 202200), is a rare autosomal recessive disorder caused by a mutation in adrenocorticotropic hormone (ACTH) receptors leading to ACTH resistance which cause isolated glucocorticoid deficiency.1,2 Clinical manifestations include hypoglycemia, seizures, skin hyperpigmentation, hyperbilirubinemia, and cholestasis.3-8
Multiple hormone deficiencies such as cortisol, growth hormone, and thyroxin are associated with congenital panhypopituitarism, leading to cholestasis in neonatal and infantile periods, as reported in the literature. Only a few cases have been reported about the possible role of isolated cortisol deficiency as a cause for infantile cholestasis.9-12
We report a 32-day-old infant with FGD1 with a novel MC2R gene variant who presented with cholestasis which progressed to acute liver failure.
Case description
A 32-days old male infant born at term 38 weeks of gestation to consanguineous parents with uneventful pregnancy with Apgar score was 8 and 9 at 1 and 5 minutes respectively with a birth weight of 2.8 kg, presented to the hepatology clinic with infantile cholestasis for investigations.
His initial assessment revealed normal anthropometric measurements, vital signs, and body system review apart from the presence of jaundice. His stool color was yellowish with dark urine.
Workup of cholestasis showed high total bilirubin (T.Bil) of 103 mmol/L, high direct bilirubin of 83 mmol/L, alanine transaminase (ALT) level of 138 U/L, aspartate transaminase (AST) level of 76 U/L, and alkaline phosphatase (ALP) of 590 U/L. He had a normal gamma-glutamyl transpeptidase (GGT) level of 28 U/L and a normal prothrombin time (PT) of 13 seconds. His serum albumin, bile acid, tandem metabolic screening, thyroid profile, renal functions, electrolytes, abdominal ultrasonography, and HIDA scan were normal. His vitamin D level was low, 18 ng/mL (normal range 30-100).
One week later, the patient developed severe bronchiolitis necessitating mechanical ventilation associated with acute liver failure and worsening cholestasis. There was a severe derangement of synthetic liver functions in the form of PT >28 seconds, and hypoalbuminemia of 22 g/L with worsening excretory functions of the liver T.Bil was 303.5 mmol/L, D.Bil was 204 mmol/L, and ALP was 849 U/L). His hepatocytes enzymes increased markedly, ALT was 3200 U/L and AST was 1420 U/L. His GGT was 30 U/L and his serum bile acids were 10 mmol/L (within normal range). A liver biopsy was requested but refused by the parents.
The condition was associated with hypoglycemia despite appropriate glucose infusion rate, metabolic acidosis, and electrolyte disturbance in the form of hyponatremia (sodium 127 mmol/L) while potassium was on the high normal limit (K 5.2 mmol/L). A full septic workup was done and came out to be negative.
Based on all the previous findings, adrenal insufficiency was highly suspected, and a stress dose of hydrocortisone and fludrocortisone was started followed by maintenance therapy with dramatic improvement of the patient’s general condition and he was weaned from the ventilator. His cholestasis and liver functions started to improve.
Hormonal examination was unfortunately requested while the patient was on replacement steroid therapy which made interpretation of results unreliable regarding the cortisol level (Table 1). The patient was discharged home with regular follow-up with the pediatric hepatology and endocrinology clinics.
Hormonal examination of the patient during Pediatric intensive care unit (PICU) admission (after giving the stress dose of cortisone).

On follow-up, the patient was thriving well with no attacks of hypoglycemia with complete resolution of cholestasis on hydrocortisone and fludrocortisone only without any other liver-supportive medications (Table 2).
Laboratory results on follow up visits.

It was of notice that ACTH level in 3 consecutive clinic visits (Table 2) was not suppressed despite cortisol therapy (23 mg/m2/day) which suggested the possibility of ACTH resistance and accordingly genetic testing for familial glucocorticoid Deficiency type1 (FGD1) was requested.
Molecular genetic analysis of the patient revealed a novel homozygous mutation c.763_764delAT (p. Met255ValfsX17) in MC2R which is compatible with the clinical diagnosis of familial glucocorticoid deficiency type1 (Figure 1).

Molecular genetic testing of the patient.
Screening of the patient’s family for the detected gene mutation revealed that both parents are heterozygous carriers for such mutation proving the autosomal recessive inheritance of the disease (Figure 2). The patient had one affected cousin who was tested and was found to be homozygous for the same mutation of our index case. The parents of the cousin were heterozygous. Other siblings of the patient and those of his affected cousin were apparently normal but were not genetically tested due to financial constrains (Figure 2).
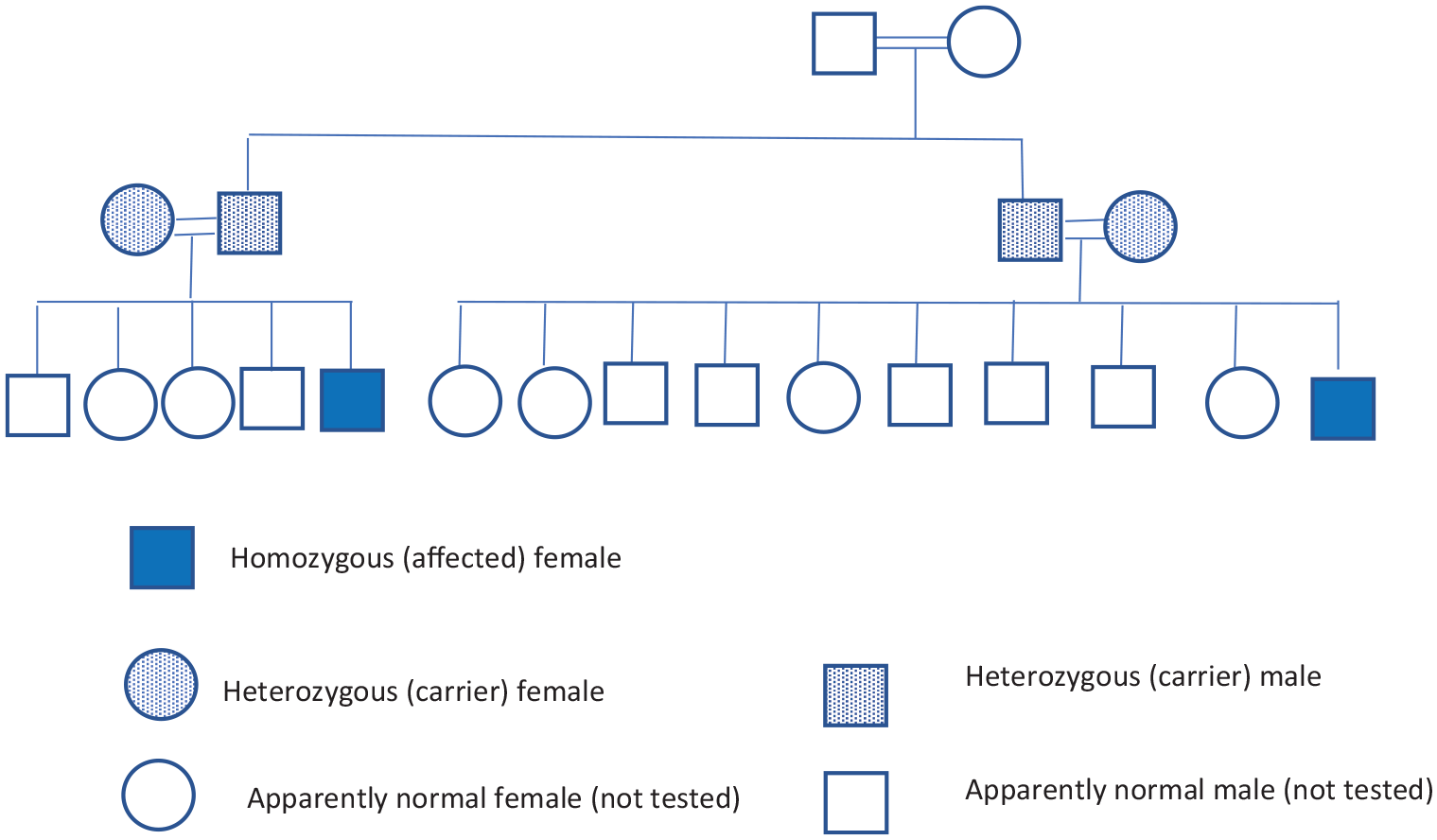
Family pedigree.
Genetic counseling about the disease nature and future pregnancies disease risk was carried for the 2 families. Parents were educated about daily disease management and sick days management protocol. The patient is currently 5 years follow-up with normal development and adequate control of his illness. His cousin is 6 months old and moved to follow in another hospital near his home with good condition.
Discussion
FGD is an autosomal recessive disorder resulting from ACTH resistance characterized by isolated glucocorticoid deficiency. Mutation in the MC2R gene account for ∼25% of patients with FGD. 1
The typical presentation is usually during the neonatal period and late childhood, with hypoglycemia, seizures, jaundice, hyperpigmentation, failure to thrive, and frequent or severe infections. 3
FGD can be easily missed, particularly in a child with acute illness. 13 Our patient presented initially with hypoglycemia, hyperpigmentation, sepsis-like picture, and prolonged cholestasis with acute liver failure. The presence of electrolytes disturbance with the previous finding suggested the diagnosis of adrenal insufficiency.
The dilemma in our case was that the PICU team started the hydrocortisone before withdrawing the hormonal examination sample, which masked the low cortisol level in the presence of the high ACTH level. The low level of 17-Hydroxy-progesterone was against the diagnosis of congenital adrenal hyperplasia.
We noticed that the ACTH level in 3 consecutive clinic visits was not suppressed even with the increment of cortisol dose, which suggested ACTH resistance, and molecular genetic analysis of MC2R for familial glucocorticoid deficiency type1 was requested.
Genomic DNA was screened for mutation in the MC2R gene. The complete coding region of exon 2 of the MC2R gene was amplified by a polymerase chain reaction and analyzed by direct sequencing. The resulting sequence data were compared with the reference sequence ID NM_000529.2. 14 Sequence analysis revealed a homozygous deletion of 2 nucleotides at position c.763_764 in exon 2 of the MC2R gene (c.763_764delAT), resulting in a premature termination codon (p. Met255ValfsX17) and subsequently in degradation of the mRNA (nonsense-mediated decay) or in a truncation of the protein. This mutation has not been described in the literature so far. Screening of family members for gene mutation revealed both parents are heterozygous carriers for c.763_764delAT (p. Met255ValfsX17) mutation in MC2R.
Prolonged neonatal cholestasis due to endocrine diseases is poorly recognized and the referral to the pediatric endocrinologist is usually delayed. Cholestasis due to hypopituitarism as well as isolated cortisol deficiency had been reported in the literature. 15
Cortisol deficiency is a treatable cause of infantile cholestasis when diagnosed early. If left untreated, it may lead to cirrhosis, portal hypertension, and later liver transplantation.12,16 Our patient had neonatal cholestasis and developed acute liver failure. His cholestasis resolved within 3 months of cortisol therapy.
Our case has many points for valuable consideration; 1. The patient’s family belongs to one of the tribes in Saudi Arabia with a black skin complexion that is why hyperpigmentation was difficult to be noticed at the neonatal period, 2. Salt losing parameters due to mineralocorticoid deficiency were not a common presentation of familial glucocorticoid deficiency, and 3. The mutation detected is a novel mutation.
Conclusions
Young infants presenting with cholestasis and documented hypoglycemia should alert pediatricians to the possibility of cortisol deficiency. In a sick child, the diagnosis of FGD can be easily missed and the measurement of serum ACTH and cortisol before starting steroid therapy is crucial for the diagnosis of FGD.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
NK: set the idea of the study and designed the study. NK, AA, EA: reviewed literature, drafted the manuscript. AB, MN, MK: critically analyzed the data. All authors reviewed and approved the manuscript for final publication.
Ethical Approval and Consent to Participate
The study was approved by the research and ethical committee of the participating hospital. The parents of the reported child signed written informed consents for participation of their child in the current study.
Consent for Publication
The parents of the reported child signed written informed consents for publication of the current study.
Availability of Data and Materials
All data and materials related to the study are included in the current manuscript.