
Abstract
Immunoglobulin G4-related systemic disease (IgG4-RSD) is a fibro-inflammatory immune condition characterized by IgG4 positive plasma cells, fibrosis, and frequently elevated serum IgG4 level. Akin to sarcoidosis, IgG4-RSD is a systemic disease with diverse organ manifestations linked by common histopathologic features. IgG4-RSD typically presents sub acutely without significant constitutional symptoms or fever. Hepatic Inflammatory pseudotumor (HIP) is a rare manifestation of IgG4-RSD composed of dense lymphoplasmacytic infiltrate and extensive fibrosis. We present an older Asian male who presented with acute onset of fever and weight loss found to have IgG4-RSD complicated by a HIP and concomitant MSSA abscess.
Case Report
A 64 year old Vietnamese male with no past medical history presented for evaluation of fevers which started in April 2020. The fevers occurred at least once daily and were associated with sweating and weight loss. Over several weeks he unintentionally lost 4 kg. After a week of fevers the patient sought care. He was a smoker but otherwise healthy and on no medications. He worked as a shrimper but had not worked since November 2019 when the season closed. Physical examination was unrevealing. Serologic testing demonstrated a mild transaminitis, elevated erythrocyte sedimentation rate, and negative viral hepatitis screen. Initial imaging with right upper quadrant ultrasonography demonstrated multiple hypoechoic lesions (Figure 1). Computer tomography (CT) of the abdomen demonstrated a 6 cm multi-cystic partially enhancing mass within the right hepatic lobe (Figure 2). He was hospitalized and a drain was percutaneously placed. Purulent debris drained which grew methicillin sensitive Staphylococcus aureus (MSSA). The patient was tested for a variety of organisms and tested negative (ie, no TB, malaria, etc.) Tissue samples obtained from the hepatic lesion also demonstrated fibro-inflammatory lesions with focally increased IgG4 positive plasma cells (Figure 3). He started intravenous antibiotics and a serum IgG4 level was elevated at 322 mg/dL (normal range: 4-86 mg/dL). He was treated with 2 months of intravenous antibiotics and monitored clinically but not placed on immunosuppressive therapy. In accordance with the comprehensive diagnostic criteria of IgG4 related disease, patient fulfilled all 3 criteria to include clinical, hematological, and histopathological. Unfortunately, patient was lost to follow up during the covid pandemic and did not follow up for repeat evaluation and proper treatment.

Ultrasound with irregular masses in the right and left lobes of the liver.
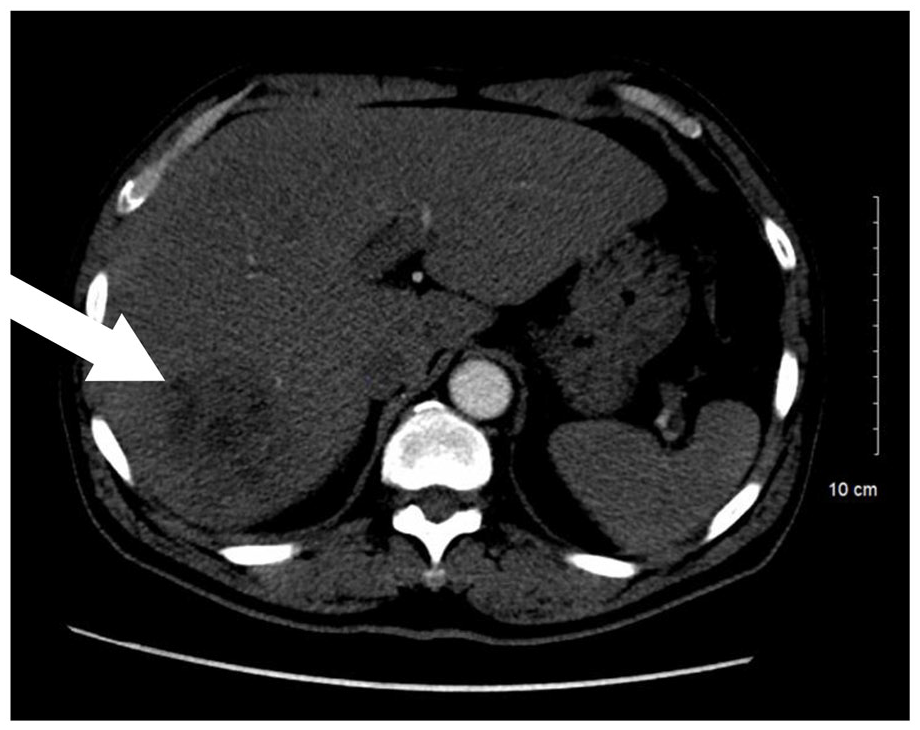
CT imaging notable for 6 cm multi-cystic partially enhancing mass (white arrow).

Tissue sample from liver biopsy demonstrating IgG4 staining plasma cells, hematoxylin, and eosin stain, 40× magnification; (right) IgG4 immunohistochemical stain, 400× magnification.
Discussion
IgG4-RSD is a fibro-inflammatory disorder recently defined as a novel clinical entity with multi-organ involvement. 1 First discovered in 1892 by Mickulcz et al in which a patient presented with symmetrical swelling of the lachrymal, submandibular, and parotid glands, and mononuclear cells on histology. 1 Thus initially it was deemed to be an atypical form of Sjogren’s disease and called Mickulcz disease. In 1967, Comings et al reported the first case of multifocal fibrosis characterized by retroperitoneal fibrosis, mediastinal fibrosis, sclerosing cholangitis, reidel’s thyroiditis, and pseudotumor of the orbit; which is currently regarded as the syndrome of IgG4-RSD.1,2 In 2011, the first international symposium on IgG4-RSD was held in Boston formally established a comprehensive and diagnostic criteria which include clinical (swelling or masses), hematological (IgG4 levels >135 mg/dL) and histopathological (marked lymphocyte, and plasmacyte infiltration and fibrosis).3,4 The criteria is scored as definite if all 3 are present, probable with only clinical and histopathological and possible with only clinical and hematological.3,4
The incidence and prevalence of the disease is difficult to estimate at this time due to its complexity and wide variety of clinical manifestations in a metachronous fashion which leads to significant under recognition. The disease is found in all ethnicities, 2:1 male predominance, median age at diagnosis is in the sixth to seventh decade of life although has been reported as early as in the 30s.4,5 In concordance with the complexity of the disease, the presentation can be variable depending on the location of the disease. Predominant hepatic lesions can present with weight loss, abdominal pain, jaundice, etc.; while cutaneously it may present as pruritic nodules, papules, and plaques but no specific characteristics have been established. Thus diagnosis can be very challenging. There are no genes currently established with the disease, however there was 1 case report which noted the disease in identical twins. 5
IgG4 related hepatopathy is becoming increasingly recognized in the literature but still remains elusive and not well characterized. 6 Hepatic inflammatory Pseudotumors (HIP) are rare manifestations of IgG4-RSD. The differential diagnosis can be difficult ranging from metastatic disease from malignancy as initially suspected in our patient, primary liver cancer or a benign lesion.7,8 The diagnosis can be also be difficult when confounded by concurrent infection with MSSA in our patient, or actinomyces reported by Lee et al. 8 HIPs have been classified into 2 major subtypes base on histological features, lymphoplasmacytic versus fibrohistiocytic. 9 The former is defined by an inflammatory process corresponding to IgG4-RSD. The 2 are histopathologically and clinically distinct with lymphoplasmacytic tumors predominantly in 60 year old males with liver dysfunction as opposed to subjective symptoms and lack of laboratory data in fibrohistiocytic tumors. As stated previously the diagnosis can be challenging with subtle clinical and laboratory findings. Laboratory data will be notable for elevated liver enzymes in a cholestatic pattern.9,10 Typically ultrasound is the first diagnostic imaging of choice which demonstrates single or multifocal hypoechoic or hyperechoic masses with increased through transmission and septation. 9 CT imaging with or without contrast notes low attenuation with several patterns of enhancement. 9 Histopathological analysis remains the cornerstone of diagnosis demonstrating a storiform pattern, obliterative phlebitis, and mild to moderate eosinophil infiltrate with positive IgG4 plasma cells.10,11
The cornerstone of treatment is derived from case reports with immunosuppressive therapy as the primary. Corticosteroids are the first line with rapid response seen in the majority of patients. However relapse is quite common after withdrawal of immunosuppression, and duration is not well established at this time. Some case reports report chronic low dose corticosteroid therapy v slow steroid taper with transition to alternative immunosuppressive agent with rituximab or rituximab monotherapy.11,12 Although therapy may be initially successful, long term risk of progressive fibrosis still remains. 12 If there is a total lack of response to immunosuppression prompt investigation should be ensued to exclude other diagnoses such as malignancy.
In conclusion, IgG4-RSD is a multi-organ fibro-inflammatory condition characterized by infiltrating lymphoplasmacytic. Clinical presentation is often subacute and without fever. Our patient was of the typical demographic for developing IgG4-RD but his presentation was atypical. Rapid onset fevers are not typical, but most likely manifested by his concomitant MSSA infection. Initial radiographic findings were concerning for infection or neoplasia, but histopathology established the diagnosis. As of mid-2020 the patient has not started any glucocorticoids or immunosuppressive therapy as he was being treated for his hepatic infection. Our case highlights certain pertinent clinical points. First, a broader definition and increased awareness is needed to allow for better understanding and improved treatment guidelines for this syndrome. Second, the complexity of establishing a diagnosis of IgG4-RD when presenting symptoms are atypical and organ infiltration may not be noted on examination until more advanced imaging is applied. Third, IgG4 related hepatic inflammatory pseudotumor is a distinct clinicopathologic disease established by histopathologic diagnosis. Finally treatment while available, is limited and outcomes can still be poor.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
The authors confirm contribution to the paper as follows: study conception and design: Francis Essien D.O., Graey Wolfley M.D, data collection: Francis Essien D.O., analysis and interpretation of results: Francis Essien D.O., Matthew Carroll M.D., draft manuscript preparation: Francis Essien D.O., Joshua Tate M.D., and Matthew Carroll M.D.
Ethical Approval
Our study did not require formal approval from an IRB board because this was a case report.
Informed Consent
Informed consent was verbally obtained from the patient and his daughter (daughter acted as translator).