
Abstract
Pycnodysostosis is a rare genetic disorder with a prevalence of 1.7 per million births; it usually presents with short stature, osteosclerosis, increased bone fragility, and acro-osteolysis of distal phalanges. There are less than 200 cases reported worldwide and very few from South-East Asia. We present a case of pycnodysostosis who presented with short stature, acro-osteolysis of distal phalanges, and on genetic testing revealing a variant c.847T>C, p.Y283H, in exon 7 of the CTSK in homozygous state: not reported till date to the best of our knowledge.
Keywords
Introduction
Pycnodysostosis, known as Maroteaux-Lamy Syndrome, was also named “Toulouse-Lautrec Syndrome” after the French artist Henri de Toulouse-Lautrec who had this disease with a prevalence of 1.7 per million.1,2 It usually presents with short stature, osteosclerosis with increased bone fragility, acro-osteolysis of the distal phalanges, and delayed closure of sutures. It is a rare autosomal recessive disorder caused by inactivating variant of the CTSK gene, which encodes for a lysosomal cysteine protease, cathepsin K. We report a case of pycnodysostosis who presented with proportionate short stature and diagnosed on genetic testing revealing an unclassified variant.
Case Report
A 4-year-old boy from Afghanistan presented to our hospital with proportionate short stature. He was the only child of parents of first-degree cousins. There was no similar illness in the family. The maternal and neonatal history was uneventful, being a home delivery; birth weight and length are not known. According to the mother, he did not feed well till 7 months of age after that he has been healthy. Developmental history was normal. Child had a prominent forehead (Figure 1), large open fontanel, and proportionate short stature with height of 93 cm (−2.46 Standard Deviation(SD)), upper-lower segment ratio 1.26, and weight 12 kg (−2.45 SD). The hands and feet had short terminal phalanges, bulbous fingertips, and broad nails. Examination of cardiovascular and respiratory systems was normal. His neurological evaluation was also normal. Abdominal examination did not reveal any hepatomegaly or splenomegaly.

Child showing dysmorphic features (frontal bossing, prominent cheeks, micrognathia, high nasal bridge, broad philtrum, and shorter fingers with spoon-shaped nails giving drumstick appearance).
All the hematological parameters were normal. Serum alkaline phosphatase, serum calcium, phosphate, vitamin D, thyroid function test, and insulin-like growth factor 1 (IGF-1) were within normal limits. Skeletal survey (Figures 2-4) shows large open fontanel, small bone island (Wormian bones), vertebrae were biconvex in the middle, and sclerotic structure of the bones in general. On the basis of clinical and radiological features, differential diagnoses of achondroplasia/hypochondroplasia and pycnodysostosis were kept in mind. Genetic testing reveals no variants in FGFR3, which ruled out the achondroplasia/hypochondroplasia.

X-ray of both hands with acro-osteolysis of the terminal phalanges with increased bone density of all the bones.
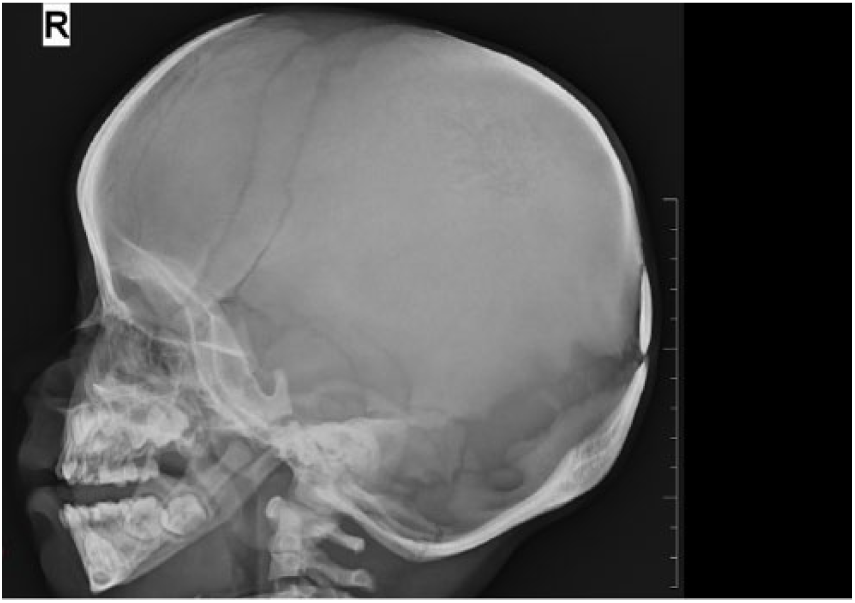
X-ray of skull lateral views showing wide open fontanel.

X-ray of spine lateral view.
Test of CTSK gene reveals an unclassified variant c.847T>C, p.(Y283H), in exon 7 of the CTSK in homozygous state. The detected variation has not been reported so far (HGMD professional database 2014.4). It alters an evolutionary conserved amino acid (all vertebrates listed in HomoloGene: 68053), and “PolyPhen” 3 predicts the functional effect of p.(Y283H) on the CTSK protein as “probably damaging” supporting the interpretation as a likely pathogenic change. “PolyPhen-2” 3 predicts the functional effect of p.(Y283H) on the CTSK protein as “possibly damaging,” and “MutationTaster” 4 calls it “disease causing.”
Discussion
Pycnodysostosis is a rare genetic disease characterized by short stature and osteosclerosis. The first case was reported in 1923 by Montanari, but this author reported his observation as an unusual clinical form of achondroplasia. 5 The incidence of this anomaly is estimated to be 1.7 per million births. Actually, less than 200 cases have been reported since the first description in 1962. 6 Twenty percent of children are born from consanguineous marriages with a male:female ratio of 2:1.
Short stature is an essential feature observed in almost all cases with a few exception of genetic variant proven cases with normal or near-normal stature. 7 It usually presents with proportionate short stature. The characteristic facial phenotype, brachydactyly, and especially bulbous terminal phalanges are diagnostic clues. As found in our case, the presence of short terminal phalanges giving rise to bulbous finger tips and broad nails is described as acro-osteolysis although there is no evidence of progressive osteolysis. Acro-osteolysis is not observed in 2 of 16 genetic variant proven children. 8
Radiological examination provides confirmatory features such as increased bone density, open fontanels, wide suture, Wormian bones, and hypoplastic clavicle. In this case, anterior fontanel was wide open with the presence of Wormian bone, and there was no hypoplasia of clavicle. Bones were osteosclerotic in general with vertebral anomalies.
Children with osteogenesis imperfecta can also have delayed closure of anterior fontanelle and short stature, but mostly short stature follows multiple fractures, leading to deformity and short limbs. Achondroplasia and hypochondroplasia are common disorders with short stature and relative macrocephaly; however, they can be easily differentiated by disproportionate short stature. The pycnodysostosis can be confused with other diseases, such as osteopetrosis and cleidocranial dysostosis, while osteopetrosis causes generalized osteosclerosis and increased bone density similar to as seen in pycnodysostosis. There is obliteration of medullary cavities in osteopetrosis; however, sparing of the medullary cavity within the long bones is characteristic of pycnodysostosis, resulting in normal hematopoietic function. 9 In cases of cleidocranial dysostosis, clavicular involvement is more severe, permitting abnormal facility in opposing the shoulders. Wide pubic symphysis, short middle phalanx of the fifth fingers, and often vertebral malformations differentiate this disorder from pycnodysostosis. Moreover, overall increased bone density as found in pycnodysostosis is not observed in cleidocranial dysplasia. 10
Short stature is the major complaint in cases of pycnodysostosis. It has been observed that a significant number of patients with pycnodysostosis have low growth hormone and IGF-1 levels and there is improvement in linear growth following growth hormone therapy. 11 , 12 However, till date, only a few studies have evaluated growth hormone response, dosages, and duration of treatment, and complications associated with growth hormone therapy are still not clear. In our patient, we have started him on growth hormone therapy; however, the patient was lost to follow-up.
Many genetic variant proven children have been reported so far from different population but very few studies from India and Afghanistan. Previous study from India also reported various novel variants suggesting the possibility of founder variants in different regions of India.13-15 Seventy percent of genetic variants reported are missense. Other reported variants from India are c.136 C>T, c.526G>T, c.577C>T, c.526G>T, c.480_481insT, p.L160fsX173, c.120+1G>T in intron 2, c.399+1G>A in intron 4, and c.148T>G (p.W50G) in exon 2; c.136C>T being the most common variant. The variant we report has never been identified before and hence is the first of its type.
Conclusion
Pycnodysostosis is a rare clinical entity with clinical and radiological features being the basics in the diagnosis of the disease. It is important to be aware of this condition as these children present to pediatricians, endocrinologist, geneticist, or orthopedic surgeons. Genetic testing is imperative in suspected cases of pycnodysostosis as not many variants have been identified in children yet. Early diagnosis will help in providing a better quality life to the patient.
Footnotes
Acknowledgements
The authors acknowledge the support of Centre of Human Genetics, Freiburg, Germany, without which this case report would have not been completed.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
IPS, AS and AA acquired and interpreted the clinical data, planned the work, wrote and revised the manuscript. All authors approved the final submitted version.
Informed Consent
Informed consent was obtained from the parents (legal guardians) for publication of the case report and accompanying images.