
Abstract
The diagnosis of maturity onset diabetes of the young (MODY) is a challenging process in view of the extensive clinical and genetic heterogeneity of the disease. Mutations in the gene encoding hepatocyte nuclear factor 1α (HNF1A) are responsible for most forms of monogenic diabetes in Northern European populations. Genetic analysis through a combination of whole exome sequencing and Sanger sequencing in three Maltese siblings and their father identified a rare duplication/frameshift mutation in exon 4 of HNF1A that lies within a known mutational hotspot in this gene. In this report, we provide the first description of an HNF1A-MODY3 phenotype in a Maltese family. The findings reported are relevant and new to a regional population, where the epidemiology of atypical diabetes has never been studied before. This report is of clinical interest as it highlights how monogenic diabetes can be misdiagnosed as either type 1, type 2, or gestational diabetes. It also reinforces the need for a better characterisation of monogenic diabetes in Mediterranean countries, particularly in island populations such as Malta with a high prevalence of diabetes.
Keywords
Introduction
Maturity onset diabetes of the young (MODY) is a rare genetically heterogenous condition characterised by beta cell dysfunction and defects in insulin secretion. Making a molecular diagnosis of MODY is challenging, in view of the multiple genes involved and the shared clinical presentation with either type 1 or type 2 diabetes mellitus (DM). A genetic diagnosis is however essential for predictive testing, proper prognostic information and personalised treatment. The molecular diagnosis of MODY has been traditionally limited using Sanger sequencing, which makes screening for mutations an expensive and time-consuming process. The increasing availability and affordability of targeted exome capture followed by high-throughput sequencing in both clinical and research laboratories has transformed molecular diagnosis, particularly in cases with atypical presentation.
In this report, we describe an frameshift mutation in exon 4 of hepatocyte nuclear factor 1α (HNF1A) with in silico predictions in favour of pathogenicity that lies within a known mutational hotspot. The mutation co-segregates with an atypical diabetes phenotype in a Maltese family and results in the loss of protein function through haploinsufficiency. HNF1A encodes a 631-amino acid transcription factor that is expressed in the liver, kidney, and pancreas. The gene is composed of 10 exons which are alternatively spliced to generate three tissue-specific isoforms. 1 In HNF1A mutations, diabetes usually develops in adolescence or early adult life, with most cases of HNF1A-MODY3 being misdiagnosed as type 1 diabetes. 2 The probands described here shared variable clinical presentations that encompassed type 1 and type 2 DM as well as presumed gestational diabetes mellitus (GDM) that failed to resolve in the post-partum period. We expand on the genetic epidemiology of MODY3 by describing the first Maltese family with an HNF1A mutation and highlight some challenges in the molecular diagnosis of atypical diabetes.
Case Report
The informed consent procedures used in this study were approved by the University of Malta institutional ethics review board (IRB 71/2013). The proband (GC) was a 30-year-old female, who was referred to diabetes clinic at 11 weeks of gestation in view of a past history of diabetes treated with insulin. At the age of 14 years, she had been diagnosed with presumed type 1 diabetes, after she had presented with osmotic symptoms of diabetes and recurrent boils. At presentation, the proband had glycosuria but no ketonuria, with a plasma glucose of 16 mmol/L, an HbA1c of 9.9%, and a body mass index (BMI) of 19.56 kg/m2. There was no diabetic retinopathy or evidence of peripheral neuropathy, and acanthosis nigricans was absent. Laboratory assays for anti-glutamic acid decarboxylase (GAD) and anti-islet cell antigen (ICA) antibodies at presentation were negative. At the time, she was started on biphasic isophane insulin and treated as probable type 1 DM. Glycaemic control was however suboptimal (HbA1c up to 8%, random glucose up to 12 mmol/L) as the proband was regularly non-compliant with her insulin therapy. She remained on insulin for approximately 8 years after diagnosis, after which she eventually stopped her treatment and spontaneously defaulted to further clinical follow-up. Interestingly, she never developed diabetic ketoacidosis. The proband eventually presented again during her pregnancy (age 30 years, 11th week gestation, primigravida) with elevated blood glucose and glycosuria, which were identified during routine antenatal evaluation. Glycaemic control throughout pregnancy was achieved through a combination of dietary measures and low-dose insulin. An elective caesarean section was eventually performed in view of polyhydramnios and foetal macrosomia, and the proband delivered a healthy infant weighing 4.6 kg.
The proband has a strong family history of diabetes. Her father (MS) was diagnosed with presumed type 2 DM at age 44 and adequate glycaemic control was achieved with a combination of a sulphonylurea and metformin. In addition, the proband has two sisters (TS and SS), both of whom presented initially with diabetes during unplanned pregnancies that however persisted after delivery. TS had presented with glycosuria at 37 weeks of gestation, aged 16 years. She required low-dose insulin during the 38th week of gestation, and hyperglycaemia persisted after delivery, requiring continued insulin therapy. The second sibling (SS) presented in a similar fashion, with glycosuria at 29 weeks of gestation aged 19 years. Low-dose insulin was required in late pregnancy and the post-partum period. No islet cell antibodies were detected in both TS and SS. The mother had no history of diabetes. Table 1 provides a summary of the clinical and biochemical characteristics of the subjects described in this report. An overview of the pedigree of this family is shown in Figure 1.
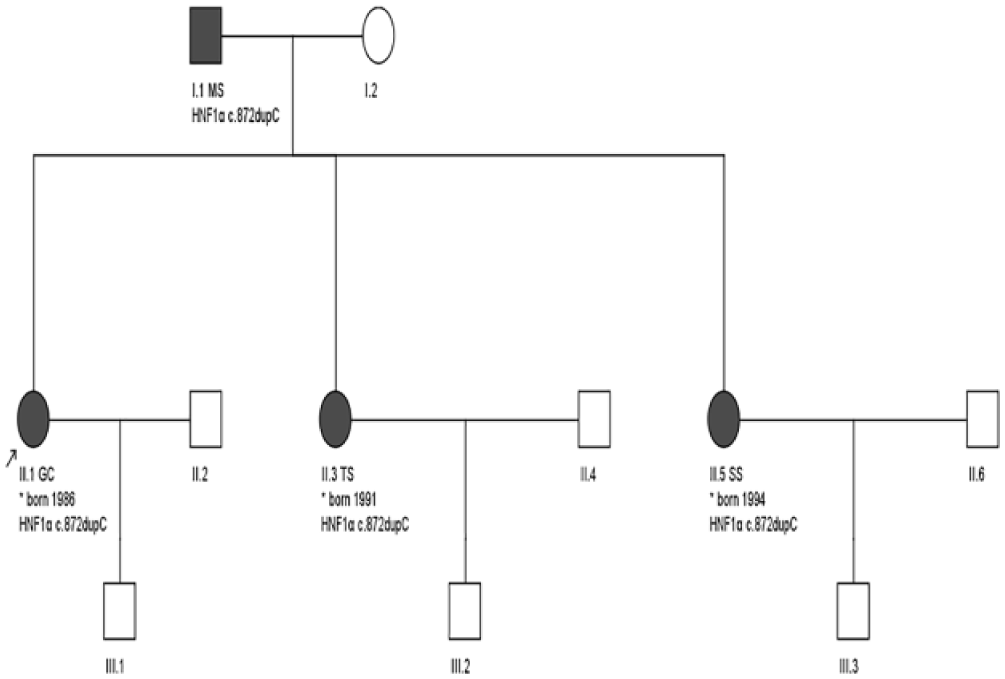
Pedigree of the family described in this study. Subjects II.1 GC and II.3 TS represent the index cases on whom whole exome sequencing was performed. Subjects I.1 MS and II.5 SS respectively represent the father and a third sibling. The c.872dupC mutation was confirmed by Sanger sequencing in these four patients, all with varying clinical presentations of diabetes. HNF1α indicates hepatocyte nuclear factor 1α.
Clinical and biochemical characteristics of the patients.

Abbreviations: BMI, body mass index; FBG, fasting blood glucose; HDL, high-density lipoprotein; LDL, low-density lipoprotein; OGTT, oral glucose tolerance test; OHA, oral hypoglycaemic agent.
Pre-pregnancy BMI.
In view of the clinical features (young age at presentation, the family history of diabetes, the absence of ketoacidosis, the variable requirement for insulin, detectable C-peptide levels, the absence of obesity, and no anti-islet antibodies), the possibility of monogenic diabetes was considered. A monogenic diabetes probability calculator showed a high probability of MODY (>75.5% positive predictive value). 3 The case was subsequently revised when the siblings and their father were offered and consented to genetic analysis for monogenic diabetes. Whole exome sequencing (WES) was performed on DNA from GC and TS, followed by targeted mutation analysis in the other family members. An overview of the sequencing and bioinformatic methodology applied is provided in the Supplementary data.
The initial search for pathogenic variants focused on the known genes for common forms of MODY. In both siblings, no pathogenic variants were detected in GCK, HNF4α, HNF1β, PDX1, INS, NEUROD1, CEL, INS, KCNJ11, or ABCC8.
In both GC and TS, a frameshift mutation at the MODY 3 (HNF1A) locus with predicted damaging effects was detected in the heterozygous state. A duplication of a C nucleotide at position 121432116 of the GRCh37/hg19 assembly was identified, corresponding to the polyC tract within exon 4 of the HNF1A gene (RefSeq accession number NM_000545.6: c.872dup) (Figure 2). This variant alters the reading frame at the glycine residue in position 292 (p.Gly292fs), resulting in premature termination of translation.

(A) Excerpt of whole exome sequencing from proband II.3 TS visualised using Integrative Genomics Viewer. The position of the pathogenic frameshift at 121432116 (hg19) in HNF1A is indicated by the vertical I bars. The mismatched base shown (C) indicates the common neighbouring single nucleotide variant, rs56348580 at position 121432117. The latter is a benign synonymous substitution c.864G>C (p.Gly288=) that was present in the homozygous form in proband II.3 TS and in the heterozygous state in the other two siblings and their father. (B) Sanger sequencing trace showing the control sequence (top) and the duplication at position 121432116 in probands II.1 GC, II.3 TS, and I.1 MS. The position of the duplication within the polyC tract in exon4 is indicated by the grey bar. The red asterisk in the trace from proband II.3 TS indicates the neighbouring benign G to C variant (rs56348580) at position 121432117, which overlaps with the peak of the C nucleotide insC mutation at 121432116. Downstream of the polyC tract, the superimposed traces caused by chromatogram shift due to the heterozygous duplication can be observed in the bottom two traces but not in the control sequence.
The polycytosine tract within exon 4 represents a known mutational hotspot within the gene. The pathogenic duplication mutation detected in this family is estimated to account for approximately 20% of HNF1A-MODY3 families. 4 In silico pathogenicity analysis of this variant was carried out using MutationTaster and CADD. The identified duplication lies in a conserved genomic region and is predicted to be disease-causing, leading to a premature termination codon and nonsense-mediated mRNA decay (probability score of 1). The Phred-scaled CADD score for this duplication is 27.7, consistent with a strong index of predicted pathogenicity for a truncating mutation in a known disease gene. The mutation is not described in the GnomAD aggregate database v2.1.
The nomenclature of indel variants in the polyC tract within exon 4 is complicated by the presence of a common G>C SNP at nucleotide position c.864, corresponding to rs56348580 (position 121432117, p.Gly288=). The latter benign variant was detected in the homozygous state in proband II.3 TS, and in the heterozygous state in father and other two siblings.
The mutation was confirmed by Sanger sequencing in both GC and TS. In addition, we sequenced exon 4 of HNF1A in the father (MS) and the third sibling (SS) and confirmed the presence of the same duplication/frameshift variant in these family members. This mutation was not detected in 300 ethnically matched unrelated controls. The family was referred back to the diabetologists for therapy review and possible sulphonylurea initiation and to the clinical genetics service for counselling. Subsequently, two siblings were successfully switched to low-dose sulphonylurea (II-3, gliclazide 80 mg bd and II-5, gliclazide 80 mg daily). No data on the index case (II-1) are available as she has been lost to follow-up.
Discussion
Whole exome sequencing of two probands with atypical presentations of diabetes revealed a duplication/frameshift mutation in exon 4 of HNF1A that results in a premature termination codon. Harries et al demonstrate that HNF1A transcripts bearing the same premature termination codon result in loss of normal protein function through haploinsufficiency as a consequence of a reduction in mRNA level through nonsense-mediated decay. 5
The same mutation was identified in two additional family members with diabetes but not in ethnically matched controls. The c.872dup mutation is classified as strongly pathogenic according to the established guidelines from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (criteria PVS1). 6
A number of null frameshift mutations in exon 4 of HNF1A that cause MODY3 have been reported in the literature. The variant identified in this family had initially been described by Yamagata et al, 7 designated P291fsinsC. Further studies had shown that this variant is the most frequently reported HNF1A mutation in MODY3 in different Northern European populations.8,9 The c.872dup p.Gly292fs mutation described here lies within the polyC tract within exon 4, which is composed of 8 or 9 cytosines (depending on the presence of the benign synonymous rs56348580 G/C SNP). Stretches of identical bases are prone to deletion or duplication of nucleotides due to slippage of the polymerase at the replication fork. 10 A review of the literature reveals examples where the commonly reported p.Pro291fsinsC mutation arises due to de novo insertions. 11
This report is significant for a number of reasons. From the clinical perspective, it highlights the scenario of monogenic diabetes presenting as hyperglycaemia during gestation in multiple family members. It also provides a striking example of the overlap in phenotype with commoner forms of diabetes and raises questions on the extent of missed diagnoses in this scenario. The report also highlights the therapeutic benefit of precision medicine in the context of atypical forms of diabetes, given that successful switch to oral hypoglycaemic agents was obtained in two siblings. It also expands on the wide array of mutations and populations that have been described for HNF1A. To our knowledge, this represents the first case report of a rare HNF1A mutation in a Maltese family.
At the clinical and population level, this report raises several issues. Primarily, the contribution of monogenic diabetes to common clinical phenotypes, such as gestational diabetes and type 2 diabetes in the high prevalence Maltese population, is as yet unknown. Several studies have investigated monogenic diabetes in pregnancy in different populations, and these have been reviewed by Colom and Corcoy. 12 Notably, studies have reported prevalence rates of GCK-MODY in gestational diabetic subjects ranging from 0.4% to 12% and up to 80%, depending on the ascertainment criteria used.13-15 The prevalence rate of HNF1A-MODY in gestational diabetes is reported to be close to 1%.16,17 In this context, the authors are presently investigating the prevalence of monogenic diabetes among Maltese cases who present with hyperglycaemia during pregnancy. Second, clinicians should be vigilant for the possibility of monogenic diabetes in young lean patients with no evidence of beta cell autoimmunity and a strong family history of the disease. Of note, the probands described in this case report were initially misdiagnosed as either type 1, type 2 DM, or GDM. Studies have shown that approximately 80% of monogenic diabetes is not diagnosed by molecular testing, reflecting either the lack of physician awareness and/or access to diagnostic service. 18 Few studies have evaluated the link between monogenic diabetes and pregnancy. Murphy 19 describes an identical case, where HNF1A-MODY3 was diagnosed in a 22-year-old lean pregnant female with presumed T1DM who defaulted to insulin therapy; while Mikuscheva et al 20 describe a case of HNF1β-MODY 5 presenting during pregnancy. A recent investigation showed that up to 6% of Danish women with diet-treated GDM have possibly pathogenic variants in GCK, HNF1A, HNF4α, HNF1β, or INS. These women are at high risk of developing diabetes after pregnancy, and the authors recommended that screening for mutations in these genes should be offered to women with GDM. 21 The management strategy of MODY3 in pregnancy reflects a balance between risk of hyperglycaemia in the first trimester with the risk of foetal macrosomia and neonatal hypoglycaemia in late gestation due to sulphonylurea therapy. 22 Rare functional variants in monogenic diabetes genes are also likely to be late-onset susceptibility variants in type 2 diabetes, further expanding the disease spectrum caused by genes traditionally associated only with highly penetrant MODY phenotypes. 23
Consequently, the authors underscore the need for a comprehensive evaluation of the genetic epidemiology of atypical non-autoimmune diabetes in the Malta. Within European populations, regional differences in MODY have been reported. Some Italian studies have reported a high prevalence of GCK-MODY in Southern European paediatric populations, while HNF1A-MODY3 is the commonest MODY subtype in Northern Europe.24,25 Interestingly, while frameshift mutations in exon 4 are reported to be prevalent in many populations, a study involving the comparable Mediterranean Greek population only detected the c.872dup variant once from total of 395 patients referred for MODY diagnosis. 26 We are presently evaluating the molecular genetics of atypical diabetes in selected Maltese families with a high probability of MODY and in separate cohort with GDM. The Maltese islands, lying between the European and African continents, have historically been subject to population bottlenecks and genetic drift, in part due to their complex history of multiple conquests, depopulation, and immigration. Several founder effects have been reported on the island.27,28 Given the high prevalence of diabetes in Malta, coupled to the reported differences in MODY across European latitudes and the high propensity for founder effects in a small island population, evaluating the molecular genetics of the disease is an important exercise with both clinical and public health implications.
Supplemental Material
supplemetary_methods – Supplemental material for Identification of an HNF1A p.Gly292fs Frameshift Mutation Presenting as Diabetes During Pregnancy in a Maltese Family
Supplemental material, supplemetary_methods for Identification of an HNF1A p.Gly292fs Frameshift Mutation Presenting as Diabetes During Pregnancy in a Maltese Family by Nikolai Paul Pace, Christopher Rizzo, Alexia Abela, Mark Gruppetta, Stephen Fava, Alex Felice and Josanne Vassallo in Clinical Medicine Insights: Case Reports
Footnotes
Acknowledgements
The authors thank the family for their collaboration in this study.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/ or publication of this article: This work was supported by institutional funds of the University of Malta.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval to the version to be published.
Availability of data and material
The dataset analysed during the current study are available from the corresponding author on reasonable request.
Informed Consent
Written informed consent was obtained from the family for publication of this case report and any accompanying images.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.