
Abstract
Cast Nephropathy/Light chain tubulopathy is usually present in patients with multiple myeloma and is very rare in patients with Waldenstrom Macroglobulinemia. There are very few case reports mentioned in the literature. We present an interesting case of Cast Nephropathy and light chain tubulopathy in an 81-year-old female patient with Waldenstrom Macroglobulinemia who required medical attention for worsening renal failure. Serum protein electrophoresis/Immunofixation showed IgM Kappa monoclonal gammopathy. Renal biopsy was remarkable for cast nephropathy and light chain tubulopathy. Furthermore on bone marrow biopsy a low grade B cell lymphoproliferative disorder with plasmacytic differentiation was present. This was most consistent with lymphoplasmacytic lymphoma, accounting for 50-60 percent of total bone marrow cellularity, in a hypercellular (60-80 percent) bone marrow.
Introduction
Waldenström macroglobulinemia (WM) is a distinct clinicopathologic entity demonstrating lymphoplasmacytic lymphoma (LPL) in the bone marrow with an Immunoglobulin M (IgM) monoclonal gammopathy in the blood. 1 Patients may present with symptoms related to the infiltration of hematopoietic tissues or the effects of monoclonal IgM in the blood.
As noted by Wang et al, 2 of the 95 797 non-Hodgkin lymphoma (NHL) cases diagnosed between 1988 and 2007 in 9 Surveillance, Epidemiology and End Results (SEER) registries, 1835 new cases of WM were recorded, representing approximately 2% of all cases of NHL. The disease is indolent, commonly affecting men (60% of the cases) and the median age at diagnosis is approximately 73 years.2,3 The clinical presentation is usually a manifestation of organ infiltration by lymphoma cells. Patients may present with anemia, lymphadenopathy, splenomegaly, neuropathy, hyperviscosity, and cryoglobulinemia. 3 WM is much more common in whites than in other ethnic groups. 4
The spectrum of kidney pathology in cases of WM and other IgM-related B-cell lymphoproliferative disorders is variable. Intracapillary monoclonal deposits, 5 amyloid deposits, light chain cast nephropathy, and cryoglobulinemic glomerulonephritis have all been reported. The non-monoclonal gammopathy-related renal pathology varies from acute tubular injury to membranoproliferative and mesangial glomerulonephritis, minimal change disease, and secondary focal segmental glomerulosclerosis.5,6 We present a rare case of light chain cast nephropathy and tubulopathy in a patient with WM.
Case Report
An 81-year-old white woman presented to the Nephrology clinic because of worsening renal function, anemia, and thrombocytopenia. Over a period of 3 months, her creatinine worsened to 3.5 mg/dL from a baseline of 1.2 mg/dL. Laboratory workup included serum protein electrophoresis and immunofixation, urine protein electrophoresis and immunofixation, all of which were suggestive of an IgM kappa monoclonal disorder. Patient’s skeletal survey was negative for any lytic lesions. Serum-free light chain assay showed significant free light chain burden with a kappa-to-lambda ratio of 27 (kappa = 69.67 mg/dL and lambda = 2.57 mg/dL). Other labs on admission showed a white blood cell (WBC) count of 6500 cells/µL, hemoglobin of 8.1 g/dL, and a platelet count of 98 000/µL.
The clinical course continued to decline and the patient developed bilateral, hemorrhagic pleural effusions for which thoracentesis was performed. In the interim, there was also mild ascites reported, which resolved spontaneously. Hemorrhagic pleural effusions were likely resulting in anemia. Abdominal computed tomography (CT) ruled out the presence of a retroperitoneal hematoma after renal biopsy. Hemolytic anemia workup was negative. Patient was finally started on plasmapheresis after consultation with oncology and received 5 procedures, with no significant improvement in her condition. Dialysis was performed for volume overload. Patient declined further treatment, decided to enter hospice care, and was deceased shortly thereafter.
Histopathology
A bone marrow biopsy was performed and showed hypercellular (60%-80%) marrow, with 50%-60% of total cellularity occupied by B cells with plasmacytic differentiation. A CD20 immunohistochemical stain was performed to confirm the presence of B lymphocytes.
Furthermore, a renal biopsy was performed to ascertain the reason for declining renal function. Morphologic evaluation of hematoxylin and eosin (and special stains including periodic acid Schiff [PAS], Jones’ methenamine silver [JMS], and trichrome)-stained biopsy sections was performed. The renal tubules showed foci of tubular injury in the form of luminal cell exfoliation, tubular dilatation, and loss of brush border and presence of proteinaceous casts. There was moderate interstitial fibrosis and tubular atrophy with 35% globally sclerosed glomeruli. The remaining glomeruli showed open capillary loops with mild mesangial expansion, but no evidence of hypercellularity. There were no capillary loop double contours, spikes and holes, or hyaline thrombi (figure 2). A Congo red stain was negative for amyloid. The presence of fibrillary glomerulopathy was also excluded based on negative DNAJB9 stain.
Immunofluorescence staining on fresh frozen tissue showed proteinaceous casts and tubular cell protein droplets that stained with kappa (3-4+), whereas it was dim to negative for lambda light chains (Figure 1A to D). Ultrastructural glomerular evaluation showed moderate podocyte epithelial foot process effacement. There were no immune-complex-mediated-type electron-dense deposits, punctate/semi-confluent (light chain type) deposits, or fibrils identified.

(A, C) Kappa immunofluorescence stain in glomerulus and tubular cell protein droplets. (B, D) The corresponding lambda light chain stain is negative.
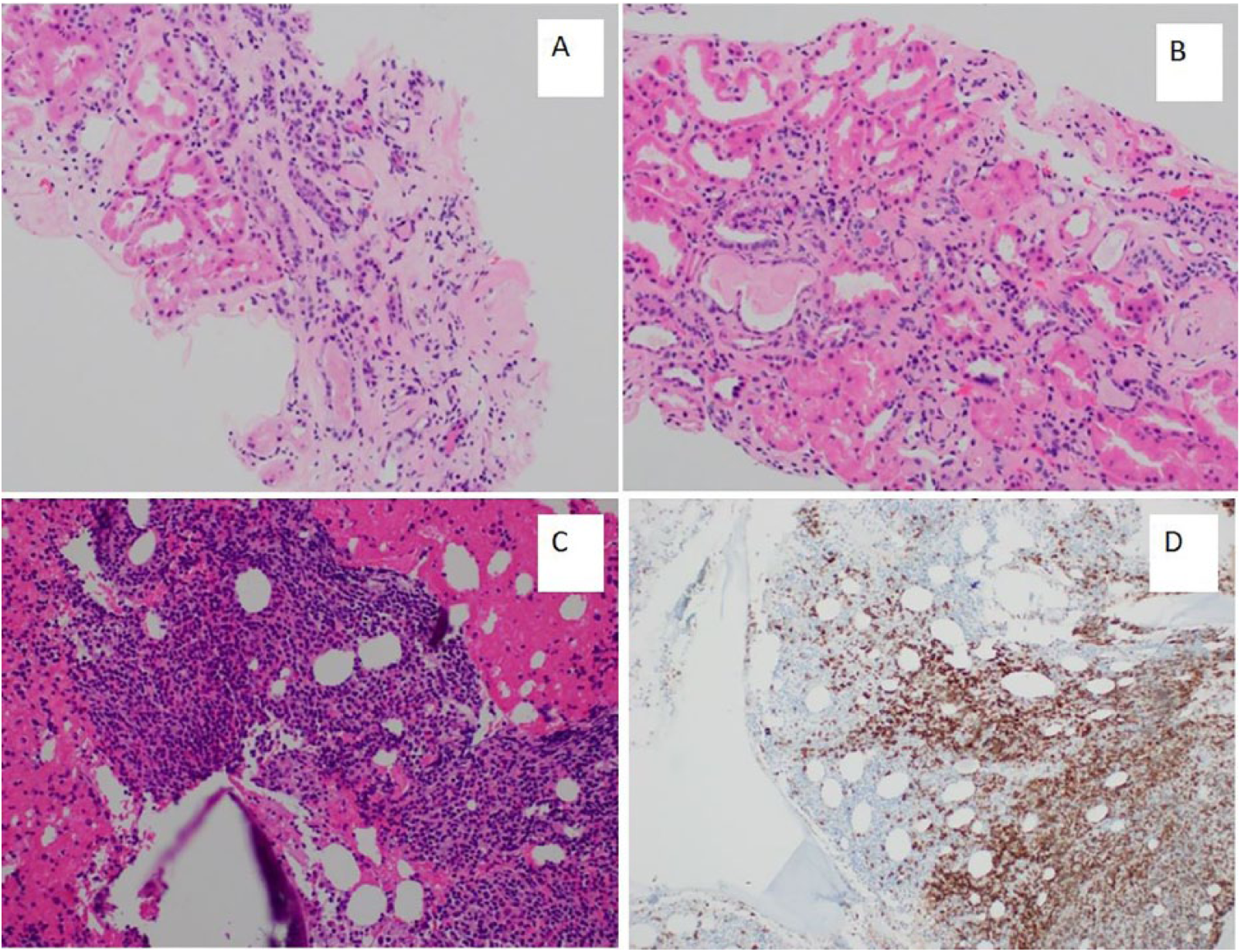
(A, B) H + E-stained sections of the renal biopsy with moderate interstitial fibrosis in the interstitium and accompanying tubular injury with casts and luminal cell exfoliation. (C) Dense lymphoid infiltrate in the bone marrow and (D) CD20 staining on the same lymphoid infiltrate highlighting the B cells.
Discussion
This case highlights an unusual presentation of WM with the presence of kappa light chain cast nephropathy and tubulopathy. No other glomerulopathy was seen on renal biopsy. The patient did not have any cryoglobulins in the blood. It has been well characterized that, under homeostasis, small amounts of free light chains are reabsorbed via scavenger receptors on the apical surface of the proximal tubular epithelium. However, in the setting of a paraproteinemia, the excess light chains exceed the capacity of proximal tubular reabsorption mechanism. In our patient, we did not see definitive crystalline inclusions by electron microscopy. Therefore, we acknowledge that there may be a co-existing component of the physiological light chain trafficking mechanism in play.
The trilineage hematopoiesis was compromised by the presence of significant neoplastic B-cell infiltration. This led to thrombocytopenia, bilateral hemorrhagic pleural effusions, and severe anemia, requiring blood transfusions. Hemorrhagic pleural effusions noted have also been described in the literature as one of the pulmonary manifestations of WM.6,7 The most frequent laboratory abnormality related to coagulation is prolongation of the thrombin time, a reflection of the inhibition of fibrin polymerization by the IgM paraprotein. 8 Our patient did not have any lytic lesions on skeletal survey, which differentiates it from a rare disorder of IgM multiple myeloma in which lytic lesions are seen. 9
Recent studies have shown that there are several genomic abnormalities that are characteristic of WM: the most common are the L265P mutation in MYD88 (found in 95%-97% of patients with WM) and a somatic mutation in CXCR4 (found in 30%-40% of patients with WM). 10 Mutation testing was not performed in our case. The patient refused aggressive intervention at the time of presentation.
In the largest series till date (57 cases) of patients with WM and other monoclonal B-cell lymphoproliferative disorders, Higgins et al 5 have described a spectrum of renal biopsy findings. These range from intracapillary monoclonal deposits to amyloid deposits. Other presentations that have been described include light chain cast nephropathy and nephrotic syndrome (commonly due to amyloid deposition), immune-mediated glomerulonephritis, which is typically due to IgM deposition or cryoglobulinemia, and non-amyloid nephrotic syndrome (with a minimal change-like picture). Although no specific lesion is identified for WM, prominent deposits of IgM in the glomerular basement membrane and infiltration of lymphocytes or plasmacytoid cells can occur. Renal insufficiency may be seen in approximately 3% of patients with WM. 5
Treatment strategy for renal manifestations of WM includes supportive treatment including hydration, plasmapheresis, and dialysis for the kidney.11,12 The clonal B-cell proliferation is addressed by combination chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab (R-CHOP). Other novel therapeutics such as bortezomib, lenalidomide, and ibrutinib may be considered on a case-by-case basis. 12 In the appropriate patient, autologous stem cell transplant can also be considered.
Conclusions
Light chain tubulopathy and cast nephropathy is usually only seen in multiple myeloma and is very rare in WM. A kidney biopsy should be judiciously pursued if patients with WM develop worsening renal function.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
MA evaluated the patient and provided clinical history and management insight. PR contributed the Pathology. Write-up was equally contributed by both authors.