
Abstract
Infiltrative heart disease (InHD) is a group of diseases characterized by the deposition of abnormal substances in the heart tissue, causing diastolic, less often systolic, dysfunction of the ventricle(s). Their classification still does not exist. In 2013, the MOGE(S) classification of cardiomyopathies was published, taking into account, along with the morphological and functional characteristics of the heart, damage to other organs, the presence of genetic mutations, acquired causes (e.g., myocardial inflammation, autoimmune diseases, storage diseases, amyloidosis), etc. By analogy with it we offer the MORAL-STAGE classification for InHD. It includes ten features: morphofunctional characteristics (M), organ damage (O), risk of cardiac death (R), age of clinical presentation, age of disease-specific therapy initiation (A), localization of the infiltrative process (inside or outside the cell, L), information about the functional class heart failure and stage of infiltrative heart disease (S), treatment (T), abnormal rhythm or conduction (A), genetic or familial nature of inheritance (G), etiology of the process (E). This article summarizes the cornerstones of the MORAL-STAGE classification and its clinical relevance. In addition, new issues are discussed that can be considered in future versions of the MORAL-STAGE classification.
Keywords
Introduction
Infiltrative heart disease (InHD) is a group of diseases characterized by the deposition of abnormal substances in the heart tissue, causing diastolic, less commonly systolic, dysfunction of the ventricle(s). InHD is a heterogeneous group of diseases, including cardiac amyloidosis, cardiac sarcoidosis, hemochromatosis, glycogen storage diseases, (Danon’s disease), Fabry disease, Wegener’s granulomatosis, Friedreich’s ataxia, mucopolysaccharidoses, cardiac oxalosis, Gaucher disease, hypereosinophilic myocardial lesions, etc.1 -4
For various diseases, classification systems provide physicians with the tools to accurately describe the pathology. Seward JB divides all InHD into 2 groups: (1) InHD, which look like hypertrophic cardiomyopathy (HCM) or hypertensive heart, and (2) InHD, which look like ischemic or non-ischemic dilated cardiomyopathy (DCM). 1 This phenotypic classification does not take into account gender, age of clinical presentation, rate of progression, the presence of genetic mutations, the risk of developing clinically apparent heart failure and sudden death, as well as the possibility of the transition of the HCM phenotype to DCM during the progression of InHD and a number of other characteristics of the disease. In addition to this classification, a universal classification of InHD has not yet been developed.
The TNM tumor staging system in oncology 5 and the MOGE(S) classification of cardiomyopathies, published in 2013, 6 prompted us to come up with the idea of the InHD classification—MORAL-STAGE system, which is described in detail below.
MORAL-STAGE Classification
The MORAL-STAGE classification takes into account 10 features (Table 1): morphological and functional phenotype (M), organ damage (O), risk of death from cardiovascular causes (R), age of clinical presentation and disease-specific therapy initiation (A), localization of the pathogenetic process (L), information about the functional status of heart failure, stage of InHD (S), treatment (T), abnormal rhythm or conduction (A), genetic mutations and inheritance pattern (G), etiology (E).
MORAL-STAGE classification.

M—Morpho-Functional Phenotype
The letter “M” corresponds to the cardiac phenotype, or morpho-functional type. The phenotype is proposed to indicate in the form of 1 letter of the corresponding form of the CM: MD (DCM), MH (HCM), MR (restrictive CM). A combination of different phenotypes is possible. HCM with a pronounced restrictive pattern is proposed to be described as MH + D. The development of dilatation as the initially hypertrophic phenotype progresses is proposed to be described as MH + D. When describing the morphofunctional phenotype, it is advisable to reflect the so-called “red flags” of amyloidosis and other infiltrative diseases, such as a short PR interval (↓PR), pathological Q wave (Q), Wolff-Parkinson-White syndrome (WPW), atrioventricular block (AVB), low QRS voltage on electrocardiography (ECG, ↓QRS). This can be displayed as MH[PR], MH[WPW] or MD[AVB], MH[↓QRS]. These red flags should be placed in square brackets after the designation of the morpho-functional phenotype. The letter E can indicate the early stage of heart damage before the formation of a certain phenotype, NS-nonspecific variant. Carriers of InHD-associated mutations without cardiac involvement are referred to as M0. When information on the cardiac phenotype is not available, such as in deceased relatives, the MNA (not appropriate) description is used.
O—Organs Involved
The letter “O” denotes organ damage. There may be an isolated lesion of the heart (OH) or a combination of damage to it and other organs, such as skeletal muscles (OH + M), auditory system (OH + A), kidneys (OH + K), nervous system (OH + N), liver (OH + Li), gastrointestinal tract (OH + G), skin (OH + C), eyes (OH + E), lungs (OH + Lu), lymph nodes (Ly). The absence of signs of damage to organs and systems is designated O0. The multiorganism of the lesion should help to suspect an infiltrative pathology.
R—Cardiovascular Mortality Risk
The letter “R” corresponds to the patient’s cardiovascular mortality risk at the moment of assessment. Risk stratification for InHD is necessary to calculate the risk of death in a particular patient, justify the need for a timely decision to start therapy. To date there has not been any widely accepted risk scale for InHD. However, for reference, we proposed 2 scales: the 5-year risk of sudden cardiac death (SCD) according to the HCM risk SCD scale in % and the 3-year risk of mortality for patients with CHF according to the MAGGIC scale in %.7,8 For example, RSCD 25, HF 19—5-year risk of SCD according to the HCM risk SCD scale of 25%, 3-year risk of mortality according to the MAGGIC scale of 19% (Tables 2 and 3).
Scale HCM-risk SCD (with modification 7 ).

MAGGIC-HF scale (modified. 8 The scale is available to users on the Internet at www.heartfailurerisk.org).

ACE, inhibitors angiotensin-converting enzyme inhibitors; ARBs, angiotensin receptor blockers; COPD, chronic obstructive pulmonary disease; SBP, systolic blood pressure.
These scales have many limitations because different diseases may present differently, but they can completely be replaced by more accurate scales in the future, or separate scales for each specific disease with similar expressions: scale name—risk (in percentage or level).
A—Age of Clinical Presentation, Age of Disease-Specific Therapy Initiation
The designation “A” includes a description of the age of clinical presentation and age of disease-specific therapy initiation. For example, A20-24 the first clinical symptoms appeared in a patient at the age of 20, and he receives disease-specific therapy since he was 24 years old. If for some reason disease-specific therapy has not been initiated, the second number may be left blank. According to these informations, clinician can understand how long the patient was on therapy, how long the delay between disease onset and treatment initiation was, and evaluate the effect of treatment depend on the age of its initiation or its duration.
L—Localization of Pathological Process Outside or Inside the Cell
The letter “L” denotes intracellular or extracellular localization of the pathological process. LO—pathological process outside cells (for example, in amyloidosis, sarcoidosis), LI—pathological process inside cells (e.g., in Fabry disease, Danon’s disease), LOI—pathological process is localized both outside and inside cells (e.g., in hemochromatosis, oxalose).
S—Stage
The letter “S” describes the stage of InHD, the stage of HF (according to ACC/AHA staging system) and the functional class of HF according to NYHA (from I to IV). By analogy with the stages of hypertension, 8 the staging of InHD should take into account the degree of damage to the heart (Figure 1):
Stage 1 (preclinical)—the stage of infiltration, the accumulation of substances in the heart begins, visualized microscopically. Patients are mostly asymptomatic and the standard clinical and imaging evaluation does not reveal any pathology. There may be an increase in specific biomarkers, initial changes on MRI and ECG.
Stage 2 (manifestations)—structural stage (pseudohypertrophy of the myocardium, thickening of the valves, valvular defects, fibrosis, dilatation of the heart chambers, etc.) and functional changes in the heart: (diastolic dysfunction, obstruction of the outflow tract of the left ventricle, etc.). Changes are detected during echocardiography, MRI, DPD scintigraphy, etc. In addition, the infiltrative process can be indirectly detected by a pathologically elevated level of highly sensitive troponin T. Clinically, this can manifest itself as unexplained chronic fatigue, decreased physical activity. The clinical picture of HF is not typical, the level of NT-proBNP, BNP is within the normal range.
Stage 3—chronic heart failure. It is characterized by fibrosis from local to diffuse. With echocardiography, structural and functional anomalies of the heart are more pronounced, incl. pseudohypertrophy and/or dilatation with impaired local contractility. Increased levels of highly sensitive troponin and NT-proBNP. Clinically, shortness of breath, rhythm and conduction disturbances are detected, in some cases requiring the installation of a pacemaker or implantation of a cardioverter-defibrillator.
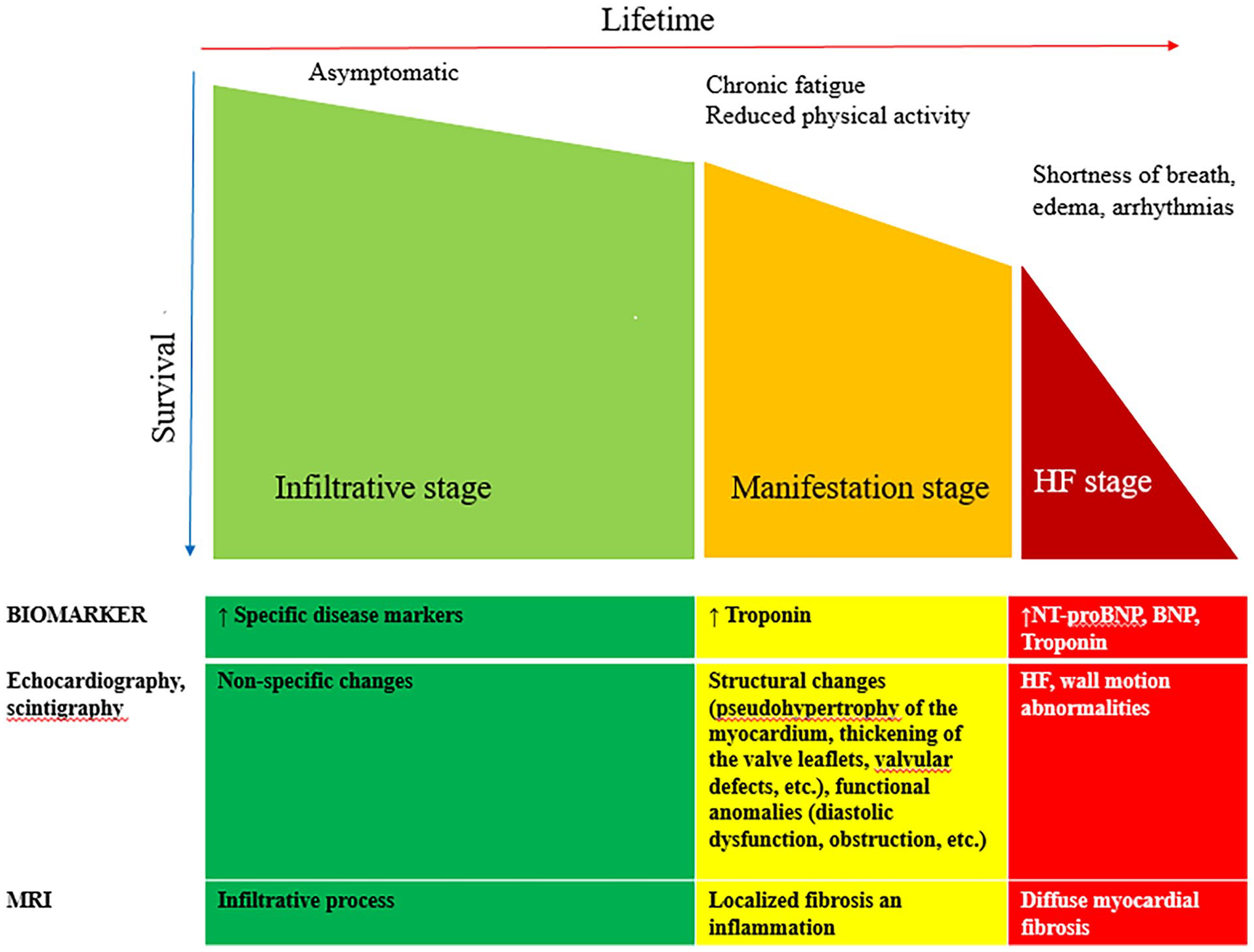
Staging of cardiac damage in patients with infiltrative heart diseases.
Understanding and identifying the 3 stages of cardiac involvement in patients with InHD is important for management. In the context of disease-specific therapy, it may appear that the long-term success of treatment is related to the stage of initiation of therapy. In addition, all of the described pathological processes and pathways may become therapeutic targets in the future.
For example, S1-A-0 denotes the first stage of InHD (infiltration stage), HF has not yet developed; S3-C-II denotes the third stage of InHD (HF stage), HF stage C according to ACC/AHA staging system, HF II according to NYHA (Figure 1).
T—Treatment
The letter “T” indicates what treatment the patient was prescribed, T0 indicates that the patient was not treated before the current visit to the doctor, TS carry out symptomatic therapy (treatment of HF, arrhythmias), you can specify a method of treatment: TS [HF]—treatment of HF, TS[ICD]—implantation of a cardioverter-defibrillator, TS[PM]—implantation of a permanent pacemaker, TS[CRT-D], TS[CRT-P]—cardiac resynchronization therapy, TDS—disease-specific therapy, after that you can specify which medicine.
A—Abnormal Rhythm or Conduction
The letter “A” indicates what type of abnormal rhythm or conduction occurs in the patient, A0—no arrhythmia, AAf—atrial fibrillation, AVT—ventricular tachycardia, etc., a combination is possible as AAf + VT—atrial fibrillation and ventricular tachycardia, AAVRT—atrioventricular reciprocal tachycardia, ALBBB—left bundle branch block.
G—Genetic
The letter “G” describes the type of inheritance, determined clinically based on pedigree analysis and genetic testing: autosomal dominant (GAD), autosomal recessive (GAR), X-linked (GXL), X-linked recessive (GXLR) or dominant (GXLD), etc. Patients with a phenotypically sporadic (GS) It is also possible to indicate a negative or unknown family history (GN or GU) and a family history not yet investigated (G0).
E—Etiology
The designation “E” includes a description of 2 points. The first testifies to the genetic (EG) or non-genetic nature of InHD. For an unidentified cause, designate EN. The second point is the designation of the exact etiology. For example, an indication of a mutation of a specific gene next to EG or an indication of the cause of the underlying disease in non-genetic InHD.
For example, in the case of familial amyloidosis (EG-ATTR [p.Val122Ile]), Fabry disease (EG-GLA [p.Trp340X]), Friedreich’s ataxia (EG-FXN), mucopolysaccharidosis (MPS) type IV (EG-GALNS). The color code assigned to each variant can provide information about the potential role of the identified variant: pathogenic or probably pathogenic (red); variant of indeterminate value (yellow); non-pathogenic variant (green). The “E” specification can describe: family members who are not carriers of the family disease-causing mutation (EG-Neg), obligate carrier (EG-OC), or obligate non-carrier (EG-ONC). EG-NA indicates that a genetic test is not available. EG-0 indicates that genetic testing was not performed or was not possible for any reason.
In non-genetic InHD, the etiology can be described as sarcoidosis (ES), non-inherited amyloidosis (EAL, EAA, etc.), secondary hemochromatosis in thalassemia (EH-T), oxalosis (EO).
Clinical Examples
Case 1
Male, 53 years old, height 178 cm, weight 86 kg, presented with dyspnea on exertion, syncope without history of myocardial infarction, diabetes, thyroid and other diagnosed disease. 9 ECG recorded AF, tachysystole, low QRS complexes voltage in the peripheral leads, right axis deviation, qR-type QRS complex (Figure 2).

Electrocardiogram of AL-amyloidosis patient. Atrial fibrillation with ventricular rate 115 per minute, northwest QRS axis, low QRS voltage at limb leads, qR at V1-4. 9
Physical examination revealed the lower extremities edema, respiratory rate 23 per minute, irregular heart rate 90 beats per minute, BP 116/68 mm Hg. Laboratory tests showed creatinine to 686 μmol/l,decrease in eGFR to 7.2, troponin I—1.020 μg/l), N-terminal precursor of brain natriuretic peptide >35,000 ng/l (Table 1). Transthoracic echocardiogram demonstrated normal ventricular function with concentric left ventricular (LV) hypertrophy without LV outflow tract obstruction (maximum left ventricular outflow gradient-11 mm Hg), significant increased LV wall thickness (up to 2.2 cm), dilatation of the left atrium (volume-98 ml, LA diameter [parasternal long axis]-43 mm), restrictive type of diastolic dysfunction and hydropericardium. Multislice computed tomography revealed bilateral hydrothorax, hydropericardium, signs of pulmonary congestion, ascites, enlargement of the liver, spleen.
The hereditary (mutant) variant of transthyretin (ATTR) amyloidosis was excluded by genetic test. The excretion of immunoglobulins light chains was inceased: kappa—63.9 mg/l, lambda—10 mg/l.
Low plasma cell count (8%) was found in the bone marrow biopsy excluded diagnosis of multiple myeloma (Table 3). Isolated amyloid deposits were found during aspiration biopsy of subcutaneous fat with Congo red staining and examination in polarized light (Figure 4). AL-type amyloid was confirmed.
Patient had chronic heart failure D stage, NYHA III
According to the MORAL-STAGE system, we can classify the patient as
Morpho-functional phenotype (M) as HCM, ECG low voltage, qR-type QRS: MH[↓R] [qR]
The patient has damage to the heart, gastrointestinal tract, liver, spleen, kidneys: OH + G + Li + Sp + K
According to the HCM risk SCD scale, the 5-year risk of SCD for the patient is 3.96%, according to the MAGGIC scale, the 3-year risk of mortality in HF is 29.2%: RSCD 3.78, HF 29.2
The age of clinical presentation was 53 years, patient died before initiation of pathogenic therapy: A61
For AL-amyloidosis, amyloid is deposited in myocardial tissues and other extracellular organs: LO
In a patient with HF stage D, III NYHA, NT-proBNP > 30,000 pg/ml, this corresponds to stage 3 of the InHD: S3-D-III
The patient was prescribed symptomatic therapy: TS [HF]
According to the ECG monitoring, AF was recorded: AAf
The patient had AL-amyloidosis (sporadic): GSEAL
In general, the patient was classified as follows
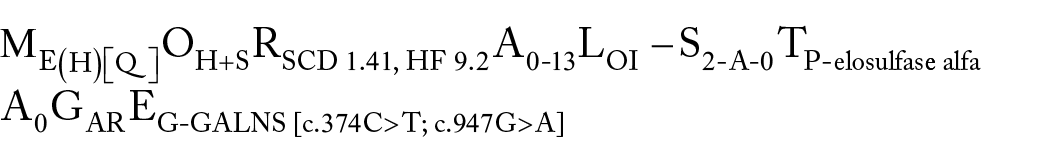
Case 2
A 63-year-old female patient admitted to the hospital with increased weakness in the distal parts of lower extremities. 10 Patient reported that from the age of 58, paresthesias and progressive weakness in the distal parts of the lower extremities began to disturb, making walking difficult. There was no history of smoking, alcohol abuse, or allergic reactions. Myocardial infarction, stroke, diabetes mellitus, arterial hypertension, lung disease denies. Initially, the symptoms were considered as manifestations of degenerative lumbar stenosis, and a decompressive laminectomy was performed at the age of 62.
Due to the persistence of symptoms after surgical treatment, she was referred to a neurological center. Based on clinical data and the results of an electroneuromyographic examination, at the age of 60, a diagnosis of axonal sensorimotor polyneuropathy was established.
Genetic testing of the patient, her older sister, son and daughter confirmed a pathogenic variant Chr18: 29178562, rs148538950, NM_000371.3:c.G368A:p. Arg123His in exon 4 of the transthyretin gene in a heterozygous state, consistent with hereditary TTR amyloidosis (hATTR) (Figure 3).

Pedigree of the patient. Women are indicated by a circle, men by a square. Family members with an identified mutation are shown in red, and unexamined family members are shown in blue (examination is planned). The numbers indicate the age.
The results of histological examination: Biopsy specimens of the subcutaneous adipose tissue of the abdomen revealed microdeposits of amyloid, grade CR 1+.
ECG showed sinus rhythm, heart rate 68 beats per minute, normal direction of the heart electrical axis and normal voltage of the QRS complex, pathological q wave in lead III.
According to Holter ECG monitoring, unstable supraventricular tachycardia, 3 episodes of ST segment depression up to 0.21 mV were registered.
Biochemical blood test: total protein 67 g/l, albumin 42 g/l, creatinine 58 µmol/l, C-reactive protein 0.9 mg/l, D-dimer 152 ng/ml, NT-proBNP 1270 pg/ml.
Echocardiography revealed concentric LV hypertrophy with a ventricular septal thickness of 13.8 mm, a posterior wall of 13.8 mm, preserved ejection fraction (55%), left atrial dilatation (46 mm, 62 mL), and pulmonary hypertension (pulmonary artery systolic pressure = 38 mmHg) and type 1 diastolic dysfunction (flow E/A = 0.76), grade 2 mitral regurgitation, grade 2 tricuspid regurgitation, grade 1 pulmonary regurgitation, no LV outflow tract obstruction.
Asymptomatic concentric cardiac hypertrophy in a patient with transthyretin familial amyloid polyneuropathy was diagnosed. The patient was recommended to start a specific anti-amyloid therapy—tafamidis (the duration of treatment is 1 year).
According to the MORAL-STAGE classification, we can classify patient as below
Morpho-functional phenotype (M) as an early stage of heart damage before the formation of the phenotype (tendency to HCM), pathological q wave in lead III on the ECG: ME(H)[Q]
The patient has damage to the heart, nervous system: OH + N
According to the HCM risk SCD scale, the 5-year risk of mortality for the patient is 2.79%, according to the MAGGIC scale, the 3-year risk of mortality is 10.2%: RSCD 2.79, HF 10.2
The age of clinical presentation was 58 years, disease-specific therapy started at the age of 62: A58-62
For ATTR-amyloidosis, amyloid is deposited in myocardial tissues and other extracellular organs: LO
The patient has no complaints, asymptomatic heart disease, LVEF 55%, NT-proBNP 1270 pg/ ml, this corresponds to stage 3 InHD: S3-B-I
The patient was prescribed long-term disease-specific therapy with tafamidis 20 mg once a day: TP-tafamidis
According to the result of Holter ECG monitoring, non-sustained ventricular tachycardia was recorded: AVT
The patient is a family member with hereditary ATTR amyloidosis: autosomal dominant inheritance pattern GAD
Molecular genetic analysis revealed the pathogenic variant c.368G > A (p. Arg123His) in the fourth exon of the transthyretin gene: EG-ATTR [p. Arg123His]
In general, the patient was classified as follows

Case 3
A 16-year-old female, height 113 cm, weight 27,7 kg, admitted to the hospital by plan for pathogenic therapy of MPS type IV. No family history of a similar condition was observed on either the maternal or paternal side.
Physical examination revealed that she had a pigeon-chest deformity, and a visible bowing of the lower extremities and normal intelligence; abdomen was distended, but no hepatosplenomegaly was observed in the patient (Figure 4). Heart rate 84 minutes. BP 100/69 mm Hg Art. A systolic murmur is heard at all points of auscultation, accent II tone.

Patient with MPS type IV: pigeon-chest deformity, distended abdomen without hepatosplenomegaly.
At the age of 3, blood and urine tests were taken for glycosaminoglycan—the activity of N-acetyl galactosamine-6-sulfatase was sharply reduced. DNA test with a complete analysis of the GALNS gene confirmed a 2 mutation: c.374C > T in exon 4, in the heterozygous state, is described in the mutation database and c.947G > A in exon 9, in the heterozygous state, is described in the database as a substitution with unknown pathogenic significance. Biochemical testing: creatinine 67 µmol/l.
MRI showed stenosis of the cervical spine, bilateral coxarthrosis, fracture of the sacrum with displacement at S4-S5 level. A slit-lamp examination did not reveal any corneal opacity.
ECG showed a sinus rhythm, moderate arrhythmia, heart rate 70-95 beats/min. Intraventricular conduction disturbance.
According to echocardiography, There is a borderline wide aortic root with asymmetry of the sinuses of Valsalva, slight LV myocardial hypertrophy (11 mm), thickening of the cusps of the valvular apparatus of the heart, combined aortic defect (aortic stenosis 1st degree, aortic regurgitation of the 1st degree). TAPSE 1.27 cm. Type 2 diastolic dysfunction. MR II. TR II-III. PR II.
The child was referred to a geneticist, and the treatment with Vimizim (BioMarin Pharmaceuticals Inc., Novato, California) (Elosulfase Alfa) was initiated at the age of 13. She had no sign of HF yet.
According to the MORAL-STAGE classification, we can classify patient as below
Morpho-functional phenotype (M) as an early stage of heart damage before the formation of the phenotype (tendency to HCM), pathological q wave in lead II, III, аVF on the ECG: ME(H)[Q]
The patient has damage to the heart, skeleton: OH+S
According to the HCM risk SCD scale, the 5-year risk of mortality for the patient is 1.41%, according to the MAGGIC scale, the 3-year risk of mortality is 9.2%: RSCD 1.41, HF 9.2
The age of clinical presentation was 0 years, disease-specific therapy started at the age of 13: A0-13
For MPS type IV, glycosaminoglycan is deposited intra- and extracellularly in myocardial tissues and other organs: LOI
The patient has no complaints, asymptomatic heart disease, LVEF 58%, BNP 52 pg/ml, this corresponds to stage 2 InHD: S2-A-0
The patient was prescribed long-term disease-specific therapy with elosulfase alfa (Vimizim) once a week: TP-elosulfase alfa
According to the result of Holter ECG monitoring, significant arrhythmia was not recorded: A0
The patient is a family member with MPS type IV: autosomal recessive inheritance pattern GAR
Molecular genetic analysis revealed confirmed a 2 mutation: c.374C > T in exon 4, in the heterozygous state, is described in the mutation database and c.947G > A in exon 9, in the heterozygous state, is described in the database as a substitution with unknown pathogenic significance of the GALNS gene: EG-GALNS [c.374C > T; c.947G > A]
In general, the patient was classified as follows

Case 4
A 53-year-old female patient, height 165 cm, weight 78 kg, with no chronic illnesses, was seen with palpitations, and progressive shortness of breath on exertion, lower limbs edema and episodes of bradycardia during night time.
She denied history of smoking and any other respiratory diseases. She was referred to the respiratory clinic with an abnormal CXR.
She had a regular tachycardia of 105 beats/min and blood pressure of 110/70 mmHg. ECG revealed a sinus tachycardia, left axis deviation and diffuse myocardial changes.
Her transthoracic echocardiogram showed abnormal segmental wall motion—hypokinesis of interventricular septum. LV morphology, systolic and diastolic function were normal. There was episodes of paroxysm of AV-node reciprocal tachycardia on her 24-hour Holter ECG.
Diagnosis of pulmonary sarcoidosis and sarcoidosis of intrathoracic lymph nodes with probable cardiac involvement was confirm by lung, lymph node biopsy and cardiac MRI.
Creatinine 68 µmol/l, SFR 89 ml/min/1.73 m2. Patient was started on corticosteroid at the age of 40.
According to the MORAL-STAGE classification, we can classify patient as below
Morpho-functional phenotype (M) as an early stage of heart damage before the formation of the phenotype: ME
The patient has damage to the heart, lung, intrathoracic lymph nodes: OH + Lu + Ly
According to the HCM risk SCD scale, the 5-year risk of mortality for the patient is 0.66%, according to the MAGGIC scale, the 3-year risk of mortality is 7.7%: RSCD 0.66, HF 7.7
The age of clinical presentation was 40 years, disease-specific therapy started at the age of 40: A40-40
For sarcoidosis, infiltrative process extracellular in myocardial tissues and other organs: LO
The patient had symptoms of chronic HF C stage, NYHA I, LVEF 62%, this corresponds to stage 3 InHD: S3-C-I
The patient was prescribed long-term disease-specific therapy with methylpred 1mg per day: TP-methylprednisolone
According to the result of Holter ECG monitoring, episodes of AVNRT was recorded: AAVNRT
The patient had sarcoidosis (sporadic): GSES
In general, the patient was classified as follows

Advantage and accessibility
Like the MOGE(S) system, MORAL-STAGE provides flexibility and can be expanded as needed. The authors believe that the nomenclature will evolve to become more complete and user-friendly as clinicians begin to put it into practice.
On first reading, MORAL-STAGE may seem like a complex nosological system, which further complicates the description of InHD. However, in practice it is quite simple to apply. The use of MORAL-STAGE does not oblige the physician to complete all fields, including genetic testing. As shown in the table, genetic tests may or may not be available. However, clinicians should make an effort to obtain a family history, especially for sudden death, and document family patterns. This system also requires the routine diagnosis of InHD in probands and relatives. Whether or not all information requested by MORAL-STAGE is immediately available does not preclude its use. In everyday practice, MORAL-STAGE can be used at the bedside and the collected data can be easily sent as a diagnosis code. In the discharge report, the final diagnosis can provide comprehensive information about the patient. The MORAL-STAGE system is a dynamic parameter that can change during follow-up.
Limitations
The current version of the MORAL STAGE classification system has a number of limitations that require future research and development. Firstly, the assessment of the risk of cardiovascular death is based on general scales for HCM and HF, there is no specific scale for InHD while this patient group has diverse etiologies. Therefore, the calculated values are for reference only and the values will be lower than the actual risk in patients. Second, the treatment section only states the type of therapy, treatment medication, and age of disease-specific therapy initiation but does not show the level of patient compliance with the treatment regimen or treatment effectiveness. Authors will research further to be able to express it more concisely and fully. Third, the presentation of information on the classification system is quite long, which can make it difficult for doctors to get acquainted. The author will find a solution so that the code letters (MORAL STAGE) and their related information have a more reasonable distribution. Hopefully in future versions the classification system will be more neat and convenient to use.
Conclusion
The dramatic increase in knowledge of InHD requires a standardized, universally acceptable classification system. The flexible MORAL-STAGE system facilitates the transition from classical medicine to the digital age of describing InHD and collects a huge amount of data that can be lost if it is not systematically recorded. The use of the MORAL-STAGE system requires a description of the results achieved at all stages of diagnosis, including clinical and cardiological examination, extracardiac examination, clinical genetics, family screening, if possible, molecular genetic analysis, morphological and histological analysis, functional status, risk stratification, as well as patient therapy. The classification provides a single language and simple, identical information to collect. It is like an individual diagnosis code for the patient, it gives the doctor a detailed idea of the disease, the patient’s condition, his approach, and also suggests the doctor further tactics.
Footnotes
Authors’ note
Thanh Luan Nguyen is also affiliated with 108 Military Central Hospital, Hanoi, Vietnam.
Author contribution
All the authors contributed significantly to the study and the article, read and approved the final version of the article before publication
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.