
Abstract
The association between hyperuricemia and cardiovascular diseases has been studied for many years. Research has shown a link between high uric acid levels and increased risk of including coronary artery disease hypertension and other cardiovascular conditions. Urate-lowering therapy, particularly with xanthine oxidase inhibitors like allopurinol, has shown promising results in reducing blood pressure in individuals with hyperuricemia and hypertension. Clinical trials and studies have demonstrated significant reductions in both systolic and diastolic blood pressure with urate-lowering treatment. Urate-lowering treatment has shown a favorable effect on reducing systolic blood pressure and major adverse cardiovascular events in patients with previous cardiovascular disease. In terms of cardiovascular safety, clinical trials have indicated that xanthine oxidase inhibitors such as febuxostat are non-inferior to allopurinol and do not increase the risk of death or serious adverse events. Overall, these findings highlight the importance of managing hyperuricemia and utilizing urate-lowering therapy to mitigate the adverse cardiovascular effects associated with elevated uric acid levels.
Keywords
Introduction
Hyperuricemia and cardiovascular diseases relationship dates back to the late 19th century. Studies conducted in the late 1800s and early 1900s found that individuals with gout, a condition characterized by elevated uric acid levels and joint inflammation, often experienced comorbidities such as hypertension, atherosclerosis, and cardiovascular events. These observations laid the foundation for further investigations into the relationship between hyperuricemia and cardiovascular diseases.1-3 Recent studies support the association of hyperuricemia and increased risk of different cardiovascular diseases, including, hypertension, heart failure, and coronary artery disease, with some showing evidence of hyperuricemia being an independent risk factor. 4
The precise mechanism of how hyperuricemia increases cardiovascular disease (CVD) risk has not been fully described, but different mechanisms have been hypothesized. Immobility, systemic inflammation, and reactive oxygen species have been proposed as culprits in multiple studies.5,6 Part of the cardiovascular regulations are mediated through the release of nitric oxide by the endothelium. Nitric oxide plays a role in vasodilation, reduction of inflammation, and inhibition of platelet aggregation. The disruption of nitric oxide production can lead to atherosclerosis development. Additionally, reactive oxygen species are also known to impair the nitric oxide production in the endothelium further increasing the risk of atherosclerosis in patients with hyperuricemia.7,8
Medication to decrease uric acid levels targets either the production of uric acid or enhances the renal excretion of uric acid. Allopurinol and febuxostat are xanthine oxidase inhibitors that decrease urate levels. With the recent association of hyperuricemia to cardiovascular events it has been hypothesized that Xanthine oxidase inhibitors could also influence the cardiovascular risk of patients with hyperuricemia.7,9 This review aims to provide an overview of the current evidence surrounding hyperuricemia and cardiovascular disease and the role of Xanthine oxidase inhibitors.
Uric Acid Synthesis
Uric acid (UA) comes from different sources, one of them being the metabolism of purines. The metabolism of adenosine and guanosine into uric acid starts with both purines in their monophosphate form, adenosine monophosphate (AMP) and Guanine monophosphate (GMP). They are then converted into inosine and guanine respectively by a series of reactions mediated by nucleotidases and deaminases. An enzyme called purine nucleosidase (PNP) converts inosine to hypoxanthine and guanosine into guanine, which is further deaminated by guanine deaminase to form xanthine. Xanthine is oxidized by xanthine oxidase becoming uric acid.10,11
Uric acid is synthesized in different organs including the liver, intestine, kidneys, muscle, and vascular endothelium. Uric acid can also be absorbed exogenously by consumption of different substances including alcohol, red meat seafood and vegetables like potatoes. It was a long thought that hyperuricemia was primarily caused by exogenous consumption of uric acid rich, however recent studies and reviews, like the one by Major et al, 12 challenged the perception while attributing greater contribution to genetic variants.12,13
The majority of urate is excreted through the kidneys with only around 30% eliminated by the gastrointestinal tract. The mechanism by which urate is eliminated from the body through the kidneys consists of filtration, reabsorption, and secretion 12 Urate is for the most part unbound plasma proteins, which means that it is filtered by the glomeruli almost in its totality. Once in the S1 segment of the proximal tubule, approximately 90 to 98% of urate gets reabsorbed. At the level of the distal proximal tubule secretion of urate occurs resulting in about 10% of all the filtered urate being excreted in about 90% being reabsorbed. 14
The normal range of uric acid levels in the blood can differ based on an individual’s gender and age. In males, the usual range for normal uric acid levels is generally between 2.5 and 7.0 mg/dL. In females, the normal range tends to be slightly lower, typically ranging from 1.5 to 6.0 mg/dL. These ranges serve as general benchmarks for the typical levels of uric acid found in the blood of healthy individuals of corresponding genders. For children, levels above 5.5 mg/dL are considered to be hyperuricemia15,16
Multiple transporters have been identified in this renal process. Urate transporter 1 (URAT1), encoded by the gene SLC22A12, serves as a urate exchanger in the apical region of the proximal tubules. It is highly involved in urate reabsorption and plays an important role in maintaining serum uric at a normal level. 17 GLUT9 encoded by the SLC2A9 gene, differs from most of the members of the GLUT for its urate rather than glucose transport and acts as a basolateral transport in the proximal tubule. Breast-cancer-resistance protein (BCRP) is encoded by the ABCG2 gene and is highly expressed in the proximal renal tubule and intestinal epithelium, was recently associated with the excretion of urate. Other transporters and factors play an important role in the balance of uric acid and its identification has represented a landmark in urate homeostasis.18,19
Since the 1960s and has been reported around 20 to 30% of your urate is eliminated daily through the gut. Notwithstanding, the mechanism by which urate is eliminated is not completely understood. Experiments have shown that the BCRP contributes also to intestinal excretion and that its polymorphisms have been associated with early onset gout in patients younger than 30 years old.17,20
Uric Acid Function
In comparison to other mammals, humans have higher uric acid levels, primarily due to the loss of uricase activity. Uricase is an enzyme that catalyzes the conversion of uric acid to allantoin, a more soluble compound that can be easily excreted. However, in humans, mutations in the uricase gene have occurred over millions of years, resulting in a decrease in uricase production. Dietary and lifestyle changes in the last century have also played a major role in the increase of average uric acid in humans from 3.5 mg/dl in the 1920s to over 6.0 mg/dl in the 1970s. When it comes to the loss of uricase activity, it is hypothesized that mutations in the uricase gene that started occurring over 15 million years ago have slowly deceased uricase production. Two different mechanism reasons for this mutation have been hypothesized. One states that mutation to the uricase promoter decreased its transcription, while the other states that mutations to the gene diminished the enzyme activity over time.21-23.
Throughout the years studies have demonstrated some of the biological activities of uric acid. It has an important role as an antioxidant, especially in the extracellular space, with many experimental studies attributing more than50% of the antioxidant function in human fluids to uric acid.17,24 Kellogg and Fridovich 25 initially described how uric acid can protect the erythrocyte lipid membrane by scavenging free oxygen radicals. This protects erythrocyte membranes from lipid oxidation when free radicals are released from reactions such as the autoxidation of hemoglobin or the production of peroxide by macrophages.25,26
Uric acid has also been proposed as an antioxidant in the central nervous system. Despite chronic elevations of uric acid being associated with an increased risk of stroke, acute elevations have been associated with lesser neurological damage in mice studies in the setting of ischemic stroke. Yu, et al 27 in an in vivo study suggested that uric acid protected the hippocampal neurons of a rat from oxidative stress. Nonetheless, in conditions such as Parkinson’s, Alzheimer’s, and Multiple Sclerosis, uric acid levels have been found to be lower. 27 It is important to point out that the anti-inflammatory properties of uric acid can be affected by the presence of key substances. For example, as demonstrated by Kuzkaya et al 28 the presence of ascorbic acid is required for uric acid to scavenge peroxynitrite, which is a powerful oxidant. The presence of bicarbonate has been shown to have the opposite effect as it impedes uric acid from preventing tyrosine nitrosylation which is an important step in the oxidative damage of proteins in the cell.26,28
The relationship between uric acid and immune pathways is still being investigated. There is evidence suggesting that uric acid may have immune-stimulating properties. Experimental studies have proposed uric acid as an immune stimulator, particularly in the context of innate immunity. Experimental studies using mice models have provided insights into the potential role of uric acid in enhancing innate immunity. These have shown that uric acid released from damaged somatic cells can serve as an enhancer of innate immunity, as it can stimulate dendritic cells and antigen-presenting cells and activate CD8 cells.28,29
The effects of uric acid on adipocytes were investigated in-vitro using an in-vitro model of adipocytes from mouse embryonic fibroblast cell line 3T3-L1. It was found that the inclusion of uric acid in the media during the differentiation process or short-term incubation with differentiated adipocytes resulted in additional generation of ROS. This effect was stopped by the superoxide scavenger MnTMPyP, which demonstrates that reactive oxygen intermediates induced by uric acid in adipocytes depend on superoxide generation. Furthermore, the effect of uric acids was found to be attenuated by the general antioxidant N-acetyl-cysteine as well as by apocynin and diphenylene-iodonium which are known inhibitors of NADPH oxidase. Uric acid was noted to induce similar effects on ROS production in human subcutaneous primary adipocytes, with MnTMPyP and apocynin attenuating this effect. The full extent of this effect is not fully understood and is not known if it only occurs on adipose tissue.26,30
Despite these known uric acid functions, the full extent of how its actions is not completely understood. Several uric acid transporters have been identified in different tissues through the years, and the complex mechanism, by which uric acid is regulated in the kidney suggests that there are other physiological functions of uric acid that have yet to be identified. 31
Hyperuricemia and Cardiovascular Diseases
Hyperuricemia and cardiovascular diseases have been linked since the latter part of the 1800s when Frederick A. Mahomed, 1 describing Bright’s disease mentioned the link between atheroma and gout. Later in 1889 Haig and Oxon revealed that arterial tension varies with the amount of uric acid circulating in the blood. 32 In the 1900s multiple reports and studies, including those by Gertler et al, 2 and Kennel et al 33 further described the association between high uric acid levels and cardiovascular disease. The former found that patients with premature coronary artery disease had higher uric acid levels than the normal population. 2 The latter stated that elevated uric acid was associated with cardiovascular disease risk in males aged 30 to 59 years based on the results of a 12-year cohort study with 5127 participants. 33
Recent studies have shown that elevated UA levels are associated with an increased risk of metabolic syndrome, hypertension, and cardiovascular diseases. 34 Morikawa et al 35 conducted the CARDIA (Coronary Artery Risk Development in Young Adults) study, analyzing the serum urate trajectories of 3563 participants over a 20-year span. Measurements were taken at baseline, 10, 15, and 20 years, revealing 3 distinct trajectories: low-stable, moderate-stable, and high-increasing. Over a median 10.6 years of follow-up, individuals in the high-increasing trajectory exhibited a 2.89-fold higher risk for cardiovascular disease (CVD) compared to those in the low-stable trajectory, indicating a significant association between the trajectory of increasing serum urate levels and an elevated risk of CVD. 35 Rahimi-Sakak et al 36 performed a meta-analysis to analyze the data of 1 134 073 participants from published studies until 2018. They found a positive association between uric acid levels and cardiovascular mortality. They reported that this association was stable with uric acid levels above 6mg/dL. 36
Hyperuricemia cutoffs are based on the risk of developing gout, nonetheless, recent studies have shown that the level of uric acid associated with cardiovascular diseases and mortality is lower. The URRAH study (Uric Acid Right for Heart Health) was a retrospective, observational multicenter study that gathered data from various large population-based longitudinal studies in Italy, as well as individuals recruited from hypertension clinics affiliated with the Italian Society of Hypertension. The study included a total of 22 714 subjects. This study revealed an independent association between serum UA levels and both total mortality (hazard ratio, 1.53 [95% CI, 1.21-1.93]) and cardiovascular mortality (CVM) (hazard ratio, 2.08 [95% CI, 1.146-2.97]; P < .001). Specific cutoff values for serum UA were identified to discriminate total mortality (4.7 mg/dL [95% CI, 4.3-5.1 mg/dL]) and CVM status (5.6 mg/dL [95% CI, 4.99-6.21 mg/dL]). 37 The ATTICA study, 38 which included 1687 healthy volunteers, identified optimal serum UA cut-off values for predicting 10-year CVD incidence as 5.05 mg/dL (0.30 mmol/l) for men and 4.15 mg/dl (0.25 mmol/l) for women.
Besides acting as a physical barrier that separates the inside cavity and the blood vessel wall, the endothelium serves as an endocrine organ. It secretes vasoactive agents such as nitric oxide (NO) and prostaglandin I2 which have vasodilatory properties. It also secretes the vasoconstrictors endothelin-1 thromboxane A2 and Angiotensin II. These substances are instrumental in regulating inflammation, oxidation, thrombosis, and vascular tone. The endothelium maintains vascular homeostasis by balancing the promotion and inhibition of growth, pro-thrombotic and anti-thrombotic activities, pro-inflammatory and anti-inflammatory responses, and pro-oxidant and anti-oxidant processes. Nitric oxide in particular plays an important role in preventing atherosclerosis by promoting vasodilation, inhibiting cell proliferation, and reducing leukocyte and platelet adhesion. Decreased NO production or increased NO inactivation can lead to reduced NO bioavailability and endothelial dysfunction.39,40
Uric acid has been shown to induce endothelial and smooth muscle cell inflammation which leads to endothelial dysfunction. Recent studies have suggested that hyperuricemia may contribute to endothelial dysfunction by reducing the bioavailability of nitric oxide. This damage is done by the ability of uric acid to generate ROS in the intracellular space. One proposed mechanism for this relationship is that hyperuricemia may cause oxidative stress and inflammation in the endothelium, leading to impaired nitric oxide production and increased degradation of nitric oxide. The effect of uric acid on reducing nitric oxide (NO) production in endothelial cells is likely due to uric acid being taken up through uric acid transporters (UATs) and exerting its effect on the intracellular space of the cells. This is supported by the fact that the reduced NO production was eliminated when UAT inhibitors were used, suggesting that the uptake of uric acid through UATs is necessary for this effect to occur 17
Hyperuricemia can lead to urate crystal deposition and chronic inflammation. This can inhibit adenosine monophosphate (AMP) associated protein kinase, which affects the innate immune responses. 41 While epidemiological studies suggest a link between hyperuricemia and cardiovascular, metabolic, and renal comorbidities, Mendelian randomization studies have not provided proof of causality. Discrepancies between observational studies and clinical trials make it difficult to develop recommendations for urate-lowering therapy in patients with asymptomatic hyperuricemia. 42
Hyperuricemia and Hypertension
Hypertension represents the primary modifiable risk factor for cardiovascular disease and overall mortality globally. As of 2010, approximately 1.39 billion adults worldwide, experienced hypertension which is defined as systolic blood pressure equal to or exceeding 140 mmHg and/or diastolic blood pressure equal to or exceeding 90 mmHg.43,44
The association between increased serum urate and hypertension has indeed been a topic of intense debate. Experimental evidence, on the other hand, strongly indicates that an increase in intracellular urate is a significant factor in the development of primary hypertension. Approximately 25% of individuals with untreated hypertension have hyperuricemia. This number is even higher in malignant hypertension where three-quarters of patients have hyperuricemia. The prevalence of hyperuricemia is higher in patients with more severe hypertension and is associated with an increased risk of uncontrolled blood pressure values and resistance to antihypertensive treatment 45
The 2018 European Society of Hypertension-European Society of Cardiology (ESH-ESC) Guidelines on Hypertension and more recent International Society of Hypertension (ISH) Guidelines on Hypertension have included serum uric acid as an additional risk factor for cardiovascular disease in the hypertensive population. These guidelines emphasize the importance of measuring uric acid levels in the presence of high blood pressure.46,47
A prospective cohort study by Ma et al, 48 which included 5758 Chinise adults between the ages of 30 and 60 years. Five distinct trajectories of UA levels were identified: low-stable, low-increasing, moderate-increasing, high-decreasing, and high-stable. Over a median follow-up period of 5.9 years, 284 women and 674 men developed hypertension. When compared to the low-stable group, the moderate-increasing group exhibited the highest risk, with adjusted hazard ratios (HRs) of 2.48 in women and 1.84 in men. The high-stable group also demonstrated increased risk, with adjusted HRs of 1.97 in women and 1.45 in men. The low-increasing group, even with UA within the normal range, displayed an elevated risk, with adjusted HRs of 1.83 in women and 1.42 in men. 48
Several animal models have been used to study the link between hyperuricemia and hypertension. While rats and mice naturally produce urate oxidase, which breaks down uric acid; in mouse knockout models, excessive hyperuricemia and kidney disease are observed, along with modest metabolic abnormalities and the development of hypertension and lipid abnormalities. One of these studies by Mazzali et al 49 the researchers found that the rats with mild hyperuricemia developed hypertension, along with early interstitial fibrosis in the kidneys. Importantly, there was a direct relationship between uric acid levels and blood pressure values in these rats. To further investigate the role of uric acid in hypertension, the researchers tested the effects of lowering uric acid levels. Results showed that both allopurinol and benzbromarone were effective in preventing the development of hypertension and kidney damage in the hyperuricemic rats.49,50
Studies on both human culture cells and animal models have shown that UA can affect the renin-angiotensin system (RAS). 51 In a study conducted by Kang et al, 52 it was demonstrated that uric acid plays a pivotal role in stimulating renin release through a macula densa-dependent mechanism. The investigation utilized an in vitro microperfused afferent arteriole-glomerular preparation to elucidate the intricate dynamics of uric acid-mediated intrarenal renin expression. 52 Furthermore, Zhang et al, 53 in a study using mouse adipose cells showed that UA caused an upregulation effect on key components of the RAS including angiotensinogen (AGT), angiotensin-converting enzyme-1 (ACE-1), renin, angiotensin type 1 receptor (AT1R), and angiotensin type 2 receptor (AT2R). This resulted in an increased secretion of angiotensin II protein and a notable elevation in reactive oxygen species (ROS) production within 3T3-L1 adipocytes. However, the introduction of RAS inhibitors, such as losartan or captopril, successfully mitigated these effects. 53 Similar findings were reported by Yu et al, 54 in a study using human umbilical vein endothelial cells (HUVECs) showed that UA induced a localized response in the RAS, leading to increased production of angiotensin II and heightened expression of its receptors, AT1R and AT2R. 54
Yang et al, 55 in an experimental study on HUVECs demonstrated that UA upregulates the Pro-renin receptor ((P)RR) which is widely expressed in endothelial cells and is able to induce RAS activation. In the study, UA exposure led to vascular inflammation with elevated cytokines and enhanced monocyte adhesion. Moreover, silencing of ((P)RR) alleviated UA-induced vascular inflammation, while Probenecid treatment effectively suppressed (P)RR up-regulation, abolishing UA-induced vascular inflammation. 55
Uric acid also has been associated with hypertension by causing afferent arteriopathy which can lead to impaired renal blood flow, which in turn can lead to renovascular hypertension. The precise mechanism is not fully known, but it has been suggested that UA induced inflammation, oxidative stress and endothelial dysregulation may play a role. 51
Hyperuricemia and Coronary Artery Disease
Gill et al 56 conducted a meta-analysis with the goal of investigating the relationship between uric acid levels and ischemic heart disease. Evidence showed that higher serum urate levels were associated with an increased risk of coronary heart disease (odds ratio, 1.2), stroke (1.1), and peripheral artery disease (1.1). Also, elevated blood pressure was estimated to mediate about one-third of the effect of urate on cardiovascular disease risk. Furthermore, urate-lowering treatment was found to favor reducing systolic blood pressure and MACE in patients with previous cardiovascular disease (odds ratio, 0.40). However, no significant effect on MACE in all individuals (odds ratio, 0.67) 56
Another meta-analysis performed by Liang et al 57 to investigate the relationship between SUA and coronary artery calcification (CAC). After analysis, 11 studies were identified involving 11,108 adults, they found that hyperuricemia is closely associated with an increased risk of CAC development and CAC progression in asymptomatic patients. The patients with high SUA levels had a 1.806-fold risk for developing CAC, and for each increase of 1 mg/dL of SUA level, the risk of CAC progression was increased by 31%. 57
Research findings have indicated a significant correlation between elevated uric acid levels and increased concentrations of NT-proBNP in individuals with suspected or established coronary artery disease (CAD) who do not exhibit overt heart failure. This association persists even after adjusting for established cardiovascular risk factors, medication usage, left ventricular ejection fraction, and other potential confounding variables. 58 Monitoring SUA levels is emerging as important in asymptomatic patients as a potential risk factor for CAC development and progression. 57
Khameneh Bagheri et al 59 conducted a study involving 100 patients diagnosed with ACS, including 59 males and 41 females. The serum level of UA was measured at the time of hospital admission, and the patients’ profiles were used to calculate the HEART score, with the average score being 6, with a range of 2 to 10. The average age of the participants was 61.37 ± 12.08 years. Among enrolled patients, the most prevalent risk factors were hypertension (48%), a history of coronary artery disease (40%), and diabetes mellitus (33%). The average serum level of uric acid was 5.81 ± 1.81 mg/dl, The study found a positive correlation between the measured UA level and the calculated HEART score, but the correlation did not reach statistical significance (R = 0.375, P = 0.090). 59
Cardiovascular Effect of Urate Lowering Agents
International and European guidelines for managing hyperuricemia recommend allopurinol as the first-line drug, with febuxostat, another xanthine oxidase inhibitor, as the second-line option. Allopurinol and its metabolite oxypurinol inhibit XO activity and are also structural analogs of hypoxanthine and xanthine, respectively, competing for XO binding. Apart from its use for gout treatment and prevention, in the past 5 years several randomized controlled trials have explored the efficacy of allopurinol in reducing cardiovascular and renal outcomes in patients with asymptomatic hyperuricemia. Although results have been mixed, some studies have shown benefits of XO inhibitors on CVD. 60
A study by MacIsaac et al, 61 utilizing data from the United Kingdom Clinical Research Practice Datalink, assessed allopurinol’s impact on cardiovascular outcomes in adults over 65 years with a history of hypertension. The study had 2032 allopurinol-exposed and 2032 nonexposed patients. Results showed that allopurinol use was associated with a significantly reduced risk of both stroke (hazard ratio, 0.50; 95% confidence interval, 0.32-0.80) and cardiac events (hazard ratio, 0.61; 95% confidence interval, 0.43-0.87), when compared to nonexposed control patients. Furthermore, in the allopurinol-exposed group, high-dose treatment (n = 1052) was associated with a lower risk of both stroke (hazard ratio, 0.58; 95% confidence interval, 0.36-0.94) and cardiac events (hazard ratio, 0.65; 95% confidence interval, 0.46-0.93) when contrasted with low-dose treatment (n = 980). 61
The ALL-HEART 62 (Allopurinol vs usual care in UK patients with ischemic heart disease) trial was conducted to investigate whether allopurinol could improve MACE in patients with ischemic heart disease. The trial involved 5937 participants aged 60 years or older with no history of gout, those patients were followed for a mean duration of 4.8 years. The results showed no significant difference MACE between the allopurinol group and the usual care group, with similar rates of occurrence in both groups. 62
Febuxostat is a non-purine selective inhibitor of both the reduced and oxidized forms of XO. Febuxostat is recommended as an alternative in patients who do not tolerate allopurinol or have refractory hyperuricemia. The effect of febuxostat on CVD has been widely debated. The CARES trial conducted by the Food and Drug Administration (FDA), showed that febuxostat had no change in the incidence of CVD relative to allopurinol, but was associated with an increase in mortality compared to allopurinol. 63 Additionally, the PRIZE trial, which had 483 with hyperuricemia but asymptomatic, showed that febuxostat had no significant changes in carotid intimal medial thickness when compared to lifestyle modifications alone. 64 Nonetheless, further sub-analysis showed that the use of febuxostat was associated with better diastolic function and decreased arterial stiffness.60,65,66
The FAST study 67 was conducted to assess the long-term cardiovascular safety of febuxostat compared with allopurinol in patients with gout. Patients were optimized for allopurinol dose toward achieving a serum urate concentration of less than0·357 mmol/l (<6 mg/dL) before being randomly assigned to continue allopurinol or start febuxostat at 80 mg/day, increasing to 120 mg/day if necessary to achieve the target serum urate concentration. The study enrolled 6128 patients with median follow-up time of 1467 days. They found that febuxostat was non-inferior to allopurinol with respect to the primary cardiovascular endpoint of composite of hospitalization for non-fatal myocardial infarction or biomarker-positive acute coronary syndrome; non-fatal stroke; or cardiovascular death. Febuxostat long-term use was not associated with an increased risk of death or serious adverse events compared with allopurinol. Nonetheless the febuxostat group showed a significant reduction in all-cause mortality. 67
The FREED clinical trial (Febuxostat for Cerebral and CaRdiorenovascular Events PrEvEntion StuDy) was reported by Kojima et al. 68 They have assessed the efficacy and safety of febuxostat compared to conventional therapy with lifestyle modification in elderly individuals with hyperuricemia. The objective of the study was to determine whether febuxostat could effectively reduce the risk of adverse clinical outcomes in patients with asymptomatic hyperuricemia, regardless of their gout status. 68 Konishi et al 69 performed a post hoc subgroup analysis to investigate the effect of febuxostat in patients with and without CVD among the 1070 patients from the FREED study who were followed for 36 months. The patients with CVD had a higher risk for the primary composite endpoint, which included cerebral, cardiovascular, and renal events and all-cause mortality, compared to those without CVD (34.2% vs 23.7%). Treatment with febuxostat reduced the incidence of the primary composite endpoint in patients with CVD (HR 0.601). In the subgroup with CVD, all-cause mortality was significantly lower in the febuxostat group than in the non-febuxostat group (HR 0.160). In other words, febuxostat can reduce the risk of cerebral, cardiovascular, and renal events as well as all-cause mortality in patients with asymptomatic hyperuricemia without gout, particularly in those with a history of CVD. 69
Uricosuric agents exert their effects by acting on renal proximal tubule transporters, leading to a reduction in the reabsorption of uric acid. Probenecid has been associated with improvement of contractility after ischemia, as demonstrated by Koch et al, 70 In vitro and in vivo experiments demonstrated that probenecid, through its activity on cellular Ca2+ levels, induces an inotropic effect without causing or exacerbating injury. 70 Probenecid was also associated with improvement in systolic and diastolic function in adults with heart failure and reduced ejection fraction (HFpEF). 71
Two randomized, placebo-controlled, double-blind, crossover trials by George et al, 72 compare the impact on endothelial function of allopurinol and probenecid compared to placebo. Each study was conducted over 1 month and focused on patients with New York Heart Association Class II–III chronic heart failure. Endothelial function was evaluated through standard forearm venous occlusion plethysmography. In the initial study, Allopurinol at 600 mg/d demonstrated a noteworthy improvement in forearm blood flow response to acetylcholine compared to both Allopurinol 300 mg/d and placebo. The percentage change in forearm blood flow (mean ± SEM) was 240.31 ± 38.19% versus 152.10 ± 18.21% versus 73.96 ± 10.29%, respectively, with a significant statistical difference (P < .001). In the second study, the uricosuric agent probenecid, administered at 1000 mg for similar urate-lowering levels, did not exhibit any observable effect on endothelial function. 72
Benzbromarone is another uricosuric agent that is not available in the United States (US) due to concerns of hepatotoxicity. A study by Nakata et al, 73 compared the effects of benzbromarone and febuxostat on endothelial function in 30 patients with hyperuricemia. Endothelial function was defined as reactive hyperemia indexes (RHI) Blood levels of asymmetric dimethylarginine (ADMA) and high-molecular-weight (HMW) adiponectin were also compared. Results revealed that benzbromarone significantly decreased uric acid levels, and HMW adiponectin and the RHI significantly increased. On the other hand febuxostat use showed a tendency to decrease uric acid levels but led to a significant decrease in RHI. Neither agent affected asymmetric dimethylarginine (ADMA) levels.60,73
Verinurad is a novel URAT1 inhibitor that also inhibits at a lesser degree OAT4 and OAT1. It has been approved in the US and Europe in combination with allopurinol, for hyperuricemia, since monotherapy carries a risk of kidney injury secondary to a marked uricosuria. 74 An ongoing clinical trial aims to evaluate the efficacy and safety of verinurad and allopurinol in patients with HFpEF and serum UA above 6 mg/dL. 75 Other Novel uricosuric agents like Lesinurad, Arhalofenate and Dotinurad currently have no evidence on cardiovascular effects. 60
In the last decades, multiple medications have gained increasing attention for their cardiovascular mortality. Few of these therapies have shown positive effects on lowering serum uric acid. Particularly sodium glucose Transporter 2 inhibitor (SGLT2i), losartan, atorvastatin, simvastatin, and fenofibrate. The QUARTZ study, which was a placebo-controlled, randomized study with 71 patients, compared the use of dapagliflozin in combination with verinurad and febuxostat to the use of only verinurad and febuxostat. The addition of dapagliflozin resulted in a substantial and statistically significant reduction in serum UA levels compared to treatment with verinurad plus febuxostat alone, lowering serum UA levels below the target level recommended by the American College of Rheumatology guidelines for gout management (⩾6.0 mg/dl [⩾354 µmol/l]) in all patients involved in the study. 76 Also, there is evidence that patients with heart failure and/or type 2 diabetes, alongside concurrent hyperuricemia, benefit from SGLT2i as it also has a synergic effect with XO inhibitors which can enhance their efficacy without the need for uricosuric drug. 60
Antihypertensive drugs like RAS inhibitors, beta-blockers, and diuretics increase serum UA levels, however, calcium channel blockers and angiotensin II receptor blockers particularly losartan have shown to reduce UA levels. 77 It has been described that losartan inhibits the URAT1, this inhibition leads to increased urinary uric acid excretion, resulting in lower serum uric acid concentrations. 78
Uric Acid Lowering Drugs Effect on Hypertension
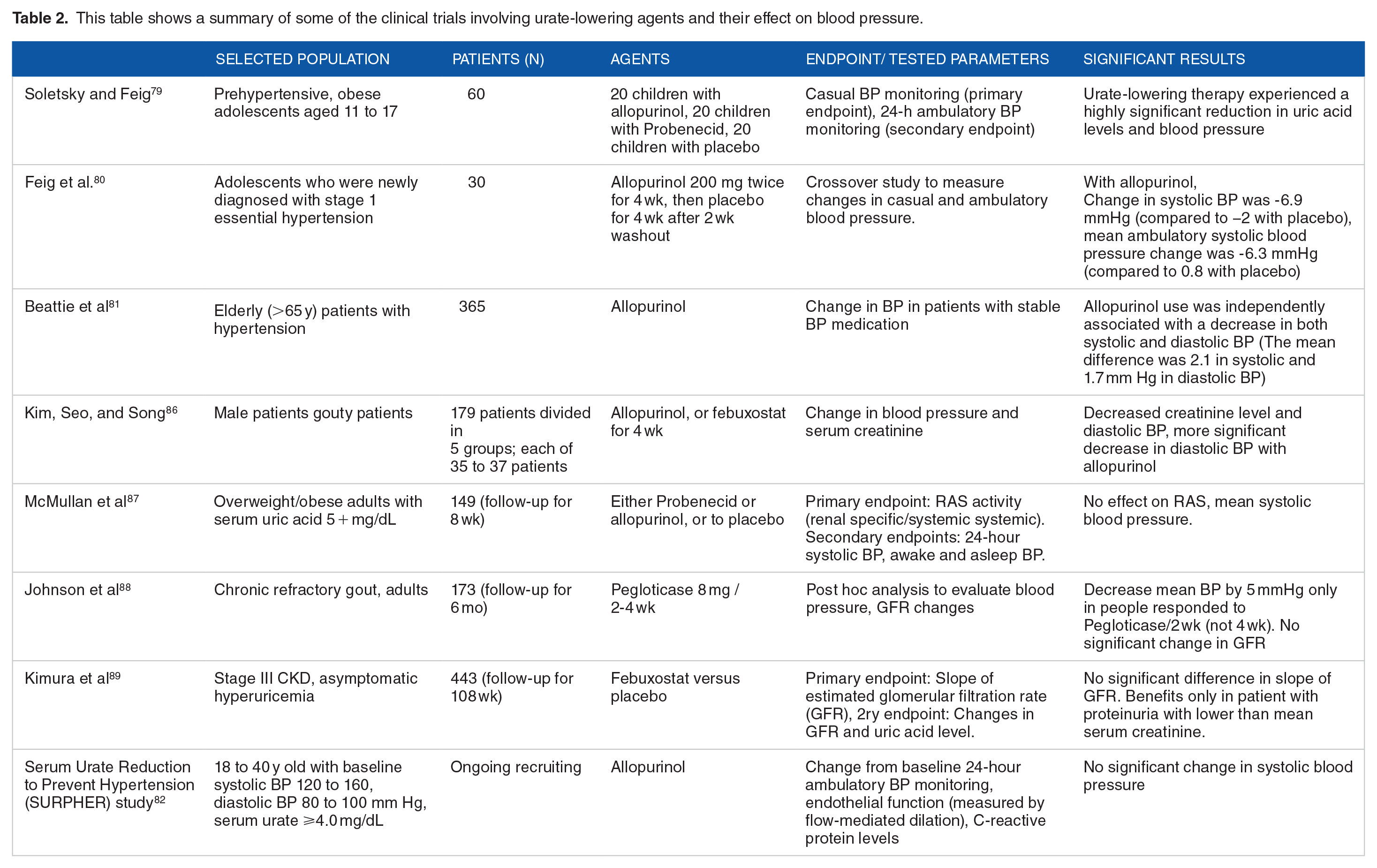
Pilot clinical trials have shown that lowering serum urate levels can have a beneficial effect on blood pressure in individuals with hyperuricemia who are young, hypertensive, and have preserved kidney function 50 Soletsky and Feig 79 conducted a study involving 60 prehypertensive, obese adolescents aged 11 to 17 years. Subjects were divided into placed in 3 different groups. one group received allopurinol, another group received probenecid, and the third group received a placebo. The study showed that subjects treated with urate-lowering therapy experienced a highly significant reduction in uric acid levels and blood pressure compared to those in the placebo group. The reduction of uric acid was measured at 2.8 mg/dL for the allopurinol group and 2.7 mg/dL for the probenecid group. In contrast, the placebo group showed only a minimal decrease of 0.3 mg/dL in serum uric acid levels. Additionally, the treatment groups experienced substantial reductions in both systolic and diastolic blood pressure. In the allopurinol group, the blood pressure decreased by approximately 10.1 mmHg systolic and 8.0 mmHg diastolic. Similarly, in the probenecid group, the systolic blood pressure decreased by approximately 10.2 mmHg systolic and 8.8 mmHg diastolic. In contrast, the placebo group showed only slight increases with rises of approximately 1.7 mmHg systolic and 1.6 mmHg, diastolic. 79
In another study conducted by Feig et al. 80 the effects of urate-lowering treatment were investigated in 30 adolescents who were newly diagnosed with stage 1 essential hypertension. The study divided patients into a treatment group with allopurinol and a placebo group. Results showed that the mean change in systolic BP with allopurinol was -6.9 mm Hg (95% CI, -4.5 to −9.3 mm Hg), whereas the change with placebo was -2.0 mm Hg (95% CI, 0.3 to −4.3 mm Hg) (P = .009). Additionally, diastolic BP was also affected by allopurinol as the mean change with allopurinol was -5.1 mm Hg (95% CI, −2.5 to -7.8 mm Hg), compared to -2.4 mm Hg (95% CI, 0.2 to −4.1 mm Hg) with placebo (P = .05). The study also evaluated mean 24-hour ambulatory BP showing that the mean change in systolic BP with allopurinol was -6.3 mm Hg (95% CI, −3.8 to -8.9 mm Hg), while it was only 0.8 mm Hg (95% CI, 3.4 to −2.9 mm Hg) with placebo (P = .001). Furthermore, the mean change in diastolic BP with allopurinol was -4.6 mm Hg (95% CI, -2.4 to −6.8 mm Hg), compared to −0.3 mm Hg (95% CI, 2.3 to −2.1 mm Hg) with placebo (P = .004). In addition to these findings, the study observed a significantly higher number of participants achieving normal BP levels, both based on casual and ambulatory criteria, while taking allopurinol (20 out of 30 participants), compared to only 1 participant while taking placebo (P < .001). 80
Studies have suggested that urate-lowering treatment can effectively reduce blood pressure in patients with an established history of HTN. Beattie et al, 81 using data from The UK Clinical Practice Research Datalink conducted a study involving 365 elderly patients with hypertension and 6678 controls. The results showed a significant reduction in BP in response to allopurinol. The mean difference between both groups was 2.1 mm Hg (95% CI, −0.6-4.8) in systolic and 1.7 mm Hg (95% CI, 0.4-3.1) in diastolic BP. Regression analysis demonstrated that allopurinol use was independently associated with a decrease in both systolic and diastolic BP (P < .001).45,81
The SURPHER Study 82 which involved a small and heterogeneous population of hypertensive patients, also compared the use of allopurinol and placebo. Results suggested that there was no significant change in systolic blood pressure during both the allopurinol treatment phase (mean ± SEM −1.39 ± 1.16 mm Hg) and the placebo treatment phase (−1.06 ± 1.08 mm Hg). Nonetheless, the study showed that endothelial function significantly improved during the allopurinol treatment periods compared to the placebo treatment periods. The increase during allopurinol treatment was 2.5 ± 0.55%, while it decreased slightly during placebo treatment (−0.1 ± 0.42%). This difference was statistically significant (P < .001).82,83
Despite several limited studies showing better blood pressure control in patients taking uric acid-lowering agents, all the studies are small-scale, leading to no clear conclusion to confirm the real benefit of uric acid-lowering agents for long-term blood pressure control. More recently, larger-scale, prospective, randomized controlled trials are debating the previously suggested positive effect of uric acid-lowering agents. More large-scale studies and subgroup interpretations would be needed to conclude the efficacy of uric acid-lowering agents in cardiovascular patients with normal uric acid levels.51,84
Conclusion
Uric acid is a metabolic waste product that is formed during the body’s breakdown of purines or from exogenous sources through the consumption of substances like red meat, seafood, some vegetables, and alcohol13,16. Over the years, researchers have observed an association between hyperuricemia and cardiovascular diseases, suggesting that hyperuricemia may act as an individual risk factor for cardiovascular disease. Observational studies have explored several potential factors that may contribute to this association. Among these factors, endothelial dysfunction, increased inflammation, and dysregulation of nitric oxide function and production have been described in observational studies. Nonetheless, the exact mechanism by which hyperuricemia increases cardiovascular risk is not yet fully understood.39-41 Establishing a definitive causal relationship between hyperuricemia and cardiovascular disease requires further investigation, including well-designed clinical trials.
There is evidence suggesting that lowering uric acid levels may have potential benefits in reducing the risk of cardiovascular disease. Although limited, xanthine oxidase inhibitors, like allopurinol and febuxostat, have shown promising results in some studies in reducing blood pressure and improving cardiovascular outcomes.69,79,80 It’s important to note that although the current evidence shows promise, more research is required to fully understand the impact of allopurinol and other xanthine oxidase inhibitors on cardiovascular outcomes. Clinical trials with larger sample sizes and longer durations are necessary to establish the effectiveness and safety of these medications. This will enable a more thorough evaluation of the benefits and potential risks associated with the use of allopurinol and other xanthine oxidase inhibitors in lowering the occurrence of cardiovascular events.
This table shows a summary of some of the clinical trials involving urate-lowering agents and their effect on cardiovascular disease.

This table shows a summary of some of the clinical trials involving urate-lowering agents and their effect on blood pressure.
Footnotes
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.