
Abstract
Systemic mastocytosis (SM) is a condition associated with clonal neoplastic proliferation of mast cells. In up to 40% of systemic mastocytosis cases, an associated clonal hematological disease of non–mast cell lineage, such as acute myeloid leukemia (AML), is diagnosed before, simultaneously with, or after the diagnosis of SM. Herein, we report a case of a 30-year-old man diagnosed with AML with inv(16)(p13;q22) CBFB:MYH11. Associated mastocytosis was not noted at diagnosis and was only detected in the bone marrow at time of remission after successful chemotherapy. The diagnosis of mastocytosis was based on the demonstration of a multifocal dense mast cell infiltrate in the marrow biopsy with aberrant immunophenotype, with coexpression of tryptase, CD117, and CD25. The mast cells showed atypical morphology mostly with irregular nuclear contour, bilobed or multilobed nuclei with cytoplasmic hypogranulation or irregular metachromatic granule distribution, and some cells with eccentric nucleus or spindle shape. Reexamination of the pretherapeutic bone marrow with immunostain for tryptase and CD25 revealed that mastocytosis was present from the start but masked by extensive blast proliferation. This case indicates that mast cell infiltrates are sometimes underappreciated at the original diagnosis of AML with inv(16) and that the concurrent diagnosis of SM with AML requires a high index of suspicion supported with comprehensive morphologic and immunohistochemical evaluation for a neoplastic mast cell proliferation.
Introduction
Mastocytosis encompasses a group of rare disorders characterized by clonal proliferation of mast cells (MC) in cutaneous and/or one or more extracutaneous visceral tissue. Based on the tissue affected, mastocytosis can be divided into cutaneous mastocytosis, systemic mastocytosis (SM), and unifocal MC tumor. The clinical course and prognosis of patients with SM range from indolent to aggressive. The 2008 World Health Organization (WHO) classification categorized a patient with SM into 4 distinct subtypes. 1 In this classification, coexistence of SM and myeloid or lymphoid hematologic neoplasm is designated as SM with associated clonal hematologic non-MC lineage disease. The name of this category has been recently shortened to “systemic mastocytosis with an associated hematological neoplasm” (SM-AHN). 2 This category represents the second most frequent subtype, comprising approximately 30% to 40% of all cases of SM.1,3
All subtypes of hematologic neoplasms have been previously reported within the context of SM-AHN. Myeloid disorders comprise 80% to 90% of the AHN, including myelodysplastic syndrome, myeloproliferative neoplasms, myelodysplastic/myeloproliferative neoplasms, and acute myeloid leukemia (AML). However, lymphoproliferative neoplasms are much less commonly implicated accounting for about 10% to 20%, with few reported cases referring to non-Hodgkin lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma, hairy cell leukemia, multiple myeloma, and acute lymphoblastic leukemia.4–7 Based on cytogenetic abnormalities, t(8;21)(RUNX1-RUNX1T1)-positive AML is the most common subtype of SM-AML. 8
The histologic and cytologic features of SM may be masked by the associated malignancy. This is especially true when the aggregates of atypical MC infiltrate are subtle, small, or infrequently present within the biopsy specimen and obscured by the non-MC proliferation. Therefore, diagnosing SM-AHN may be challenging to pathologists. Here, we report a case of AML with inv(16) in whom a coexisting mastocytosis was not detectable at initial diagnosis because of an extensive involvement by AML blasts, obviously masking the compact MC infiltrate.
Case Report
A 30-year old Nepalese man presented to emergency department with 2 weeks’ history of fever, abdominal pain, and fatigue. Physical examination revealed fever, pallor, jaundice, and hepatosplenomegaly. Peripheral blood analysis revealed moderate normochromic normocytic anemia (hemoglobin, 9 g/dL; normal, 13-17 g/dL), severe thrombocytopenia (platelets, 25 × 109/L; normal, 150-400 × 109/L), and marked leukocytosis of 164 × 109/L (normal, 4-10 × 109/L). Peripheral blood smear showed many blast cells with left shift and increased monocytic cells with a differential count of neutrophils 7%, lymphocytes 3%, eosinophils 1%, monocytes 16%, promyelocytes 8%, promonocytes 23%, and blasts 42%. The blasts were medium to large in size with fine chromatin and prominent nucleoli, some with irregular/convoluted nuclear contour (Figure 1A).

(A) Peripheral blood smear shows leukocytosis with circulating blasts (thin arrow), promonocytes (thick arrow), monocytes, and dysplastic granulocytic cells (hyposegmented neutrophils, arrow head). Inset shows eosinophilic myelocytes with abnormal coarse basophilic primary granules. (B) Bone marrow aspirate smear showing blasts cells, promonocytes, monocytes, and granulocytic cells with dysplastic features (hyposegmented neutrophils) (Wright stain, original magnification ×1000). (C) Bone marrow biopsy shows infiltration with leukemic cells, many with irregular convoluted nuclear contour with prominent eosinophilic cells (hematoxylin-eosin, original magnification ×600). Inset: Low-power bone marrow biopsy shows remarkably hypercellular marrow with extensive diffuse infiltration (hematoxylin-eosin, original magnification ×40).
Initial bone marrow (BM) aspirate stained with Wright stain was hypercellular with 39% blasts, 13% promonocytes (blasts equivalent), and increased monocytes (17%). No Auer rods were noted. Maturing myeloid cells comprised (28%) severe dysplastic features and included 2% eosinophils (Figure 1B). Bone marrow biopsy showed markedly hypercellular BM with diffuse infiltration by blasts (Figure 1C), positive for CD34, lysozyme, myeloperoxidase, and CD68, with partial positivity for CD117. There were prominent eosinophilic cells, scattered and in small groups with marked suppression of normal trilineage hemopoiesis.
Multicolor flow cytometry (FCM) analysis was performed on BM aspirate using CD45-gating strategy to identify the immunophenotype of the blasts. Acute leukemia panel of 28 antibodies in a 4-color combination (FITC/PE/ECD/PC5 fluorescent conjugates) was used, as follows: (1) CD34/CD117/CD45/CD19, (2) CD14/CD13/CD45/CD64, (3) HLA-DR/CD7/CD45/CD5, (4) CD34/CD33/CD45/CD56, (5) CD19/CD10/CD45/CD3, (6) CD15/CD33/CD45/CD2, (7) CD9/CD19/CD45/CD4, (8) CD20/CD10/CD19/CD45, (9) cMPO/cCD79a/cCD3/sCD45, (10)TdT/sCD19/sCD3/sCD45, (11) CD36/CD11c/CD45/CD11b, and (12) CD41/glycophorin A/CD45/CD61(PC7).
Flow cytometry analysis revealed approximately 56% myeloid blasts with moderate CD45 and expressing CD34, cMPO, CD33, CD13, and CD9, and the majority was positive for CD117 and HLA-DR, with partial expression of CD15 (on approximately 35% of the cells) (Figure 2). The blasts were negative for CD56, CD14, CD64, CD11b, CD11c, CD36, CD61, CD41, glycophorin A, TdT, and B-cell and T-cell markers. Monocytic cells increased (23%) expressing CD64, and the majority was positive for CD14. Cells in the granulocytic gate comprised approximately 17% showing loss of CD10 expression (features of dysmaturation).

Flow cytometry analysis of bone marrow aspirate at presentation revealed approximately 56% blast cells (green gate). Blasts are positive for cMPO, CD117, CD13, CD33, and CD34, and the majority is positive for HLA-DR. FITC indicates fluorescein isothiocyanate.
The morphology and FCM findings were consistent with the diagnosis of AML with increased monocytic cells. Cytogenetic analysis by fluorescent in situ hybridization (FISH) using a probe for CBFB (BAR) gene revealed an abnormal hybridization signal pattern, indicating rearrangement in 56% of the cells. This was confirmed by conventional karyotype which showed inv(16)(p13.1q22), concluding the diagnosis of AML with inv(16)(p13.1;q22) CBFB:MYH11 (Figure 3).

Interphase FISH and cytogenetic studies showing an inv(16). (A) Interphase FISH; CBFB Break Apart FISH Probes are used to detect inv(16); centromeric end 5′ is labeled with spectrum orange and telomeric end 3′ is labeled with spectrum green. The normal homologue of chromosome 16 shows a fusion (yellow), the abnormal chromosome 16, with inversion shown in orange and green signals apart from each other. (B) Partial karyotype shows normal chromosome 16 and inv(16) indicated with arrow. FISH indicates fluorescent in situ hybridization.
Patient initial laboratory workup revealed elevated total bilirubin of 82.6 µmol/L (normal, 3-20 µmol/L), mainly direct bilirubin of 55.8 µmol/L (normal, 0-8.6 µmol/L), and high liver enzymes, with alanine transaminase (ALT) of 187 IU/L (normal, 0-40 IU/L), aspartate transaminase (AST) of 87 IU/L (normal, 0-37 IU/L), and alkaline phosphatase of 378 IU/L (normal, 40-150 IU/L). Renal function tests were normal with creatinine of 94 µmol/L (normal, 70-115 µmol/L). Hepatitis serology screening showed hepatitis E virus IgG and IgM with evidence of past hepatitis B virus infection, for which lamivudine was given before starting chemotherapy. Magnetic resonance imaging of the liver and magnetic resonance cholangiopancreatography done to exclude biliary obstruction revealed hepatomegaly with no focal lesion, multiple porta hepatis, peripancreatic, mesenteric, and para-aortic lymph nodes with no evidence of biliary obstruction. All septic workup and quantiferon testing proved negative.
The patient was started on cytarabine (100 mg/m2) for 7 days. Anthracycline was omitted from the first induction because of elevated direct bilirubin. The patient had persistent fever during the neutropenia phase despite treatment with broad-spectrum antimicrobial and antifungal drugs. Miliary tuberculosis (TB) was suspected, for which liver biopsy was performed and was consistent with cholestatic hepatitis with the absence of granulomas or leukemic infiltration. The patient had no clinical findings, suggesting central nervous system (CNS) involvement; however, intrathecal chemotherapy was planned upfront as the patient was considered to be at high risk due to CNS involvement. Examination of cerebrospinal fluid was delayed as the patient was critically ill; it was done just before the first consolidation and revealed few blasts confirmed by FCM analysis. The patient was treated with triple intrathecal chemotherapy twice weekly for total of 8 doses.
Bone marrow examination post first chemotherapy cycle showed cellular aspirate with approximately 19% blasts, and BM biopsy showed interstitially increased primitive cells. Immunohistochemical stains showed increased CD34 positivity roughly estimated at 20% to 30% and overall increased positivity for CD68 with focal collections of CD117-positive cells interpreted as persistence of the leukemic process. After recovery of neutropenia, the patient was started on second induction 3+7 protocol. The hospital course was complicated with massive pleural effusion. Therapeutic pleural tapping was done and showed white blood count of 1000/µL with 65% lymphocytes, no blast cells, glucose of 6.2 mmol/L, protein of 50.6 g/L, lactate dehydrogenase of 104 IU/L, pH 7.5, and negative for acid-fast bacilli and TB by polymerase chain reaction (PCR). The patient was treated empirically with anti-TB drugs and steroids. Few days later, fever subsided, jaundice resolved, and the patient’s condition improved.
Evaluation of BM after second induction showed cellular BM with 3% blasts, indicating remission. However, the BM aspirate showed the presence of morphologically atypical MC comprising approximately 2% of the total cells, mostly with irregular nuclear contour (kidney-shaped, bilobed, or multilobed nuclei), pale cytoplasm with cytoplasmic hypogranulation or irregular metachromatic granule distribution, some with fine nuclear chromatin (promastocyte morphology), together with some cells showing eccentric nucleus or spindle shape (Figure 4A and B). Examination of the BM biopsy revealed multiple perivascular and randomly distributed focal collections of MC (in clusters of >15 cells) with some interstitially increased MC with spindle forms and some with lobed nucleus. Eosinophils were prominent around few of the MC aggregates. The MC were positive for tryptase, CD117, CD68, and CD25. CD2 immunostain appeared negative (Figure 5A to C). These findings established the diagnosis of SM with AML. When reexamining the BM at diagnosis, MC were not impressive in the aspirate; however, re-evaluation of the marrow biopsy with the aid of tryptase and CD25 immunostain revealed many scattered and multiple dense clusters of cells positive for CD117, tryptase, and CD25, indicating that mastocytosis was present from the start but masked by the extensive infiltration by blasts.
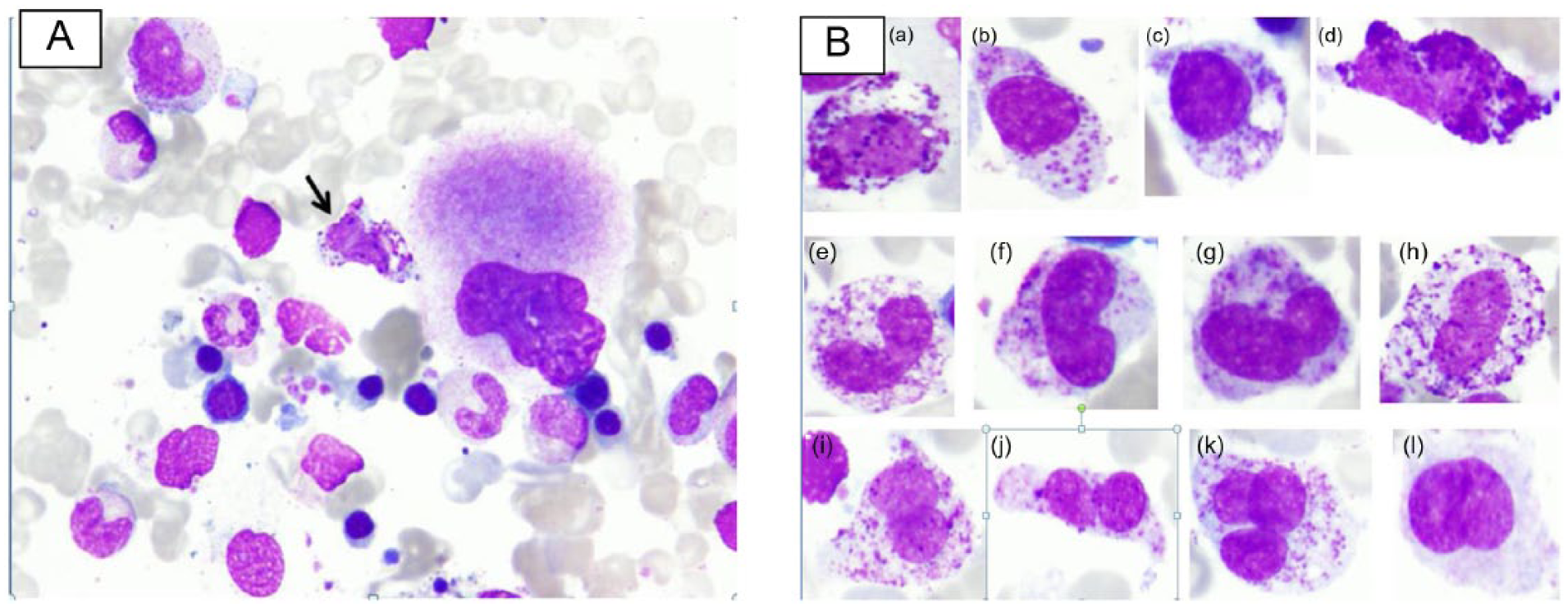
(A). Bone marrow aspirate smear shows different stages of granulocytic cells, normoblasts, and mast cells (arrowed) (Wright stain, original magnification ×1000). (B) Cytologic abnormalities of mast cells. (a to c), Atypical mast cell type 1 with eccentric nucleus and hypogranular cytoplasm; (d) atypical mast cell type 1, spindle form; (e to h) atypical mast cell type II with kidney shape, bilobed nuclei with mature chromatin and hypogranular cytoplasm; (i to k) atypical mast cell type II with bilobed or multilobed nuclei with hypogranular cytoplasm and more fine immature chromatin (promastocyte); (l) atypical mast cell type II with fine chromatin and agranular cytoplasm (Wright stain, original magnification ×1000.

(A) Bone marrow biopsy shows perivascular collections of mast cells (circled) (hematoxylin-eosin, original magnification ×100). Inset: Many of the mast cells are of elongated spindle form (hematoxylin-eosin, original magnification ×1000). (B) Bone marrow biopsy shows intertrabecular aggregate of mast cells (hematoxylin-eosin, original magnification ×400). Inset: Some of the mast cells show kidney-shaped nucleus (hematoxylin-eosin, original magnification ×1000. (C) Immunohistochemistry on bone marrow biopsy. The mast cells are positive for CD117, tryptase, and CD25, with no significant staining for CD2 (×200).
Serum tryptase was elevated at 38.5 µg/L (negative <11 µg/L), and allele-specific oligonucleotide polymerase chain reaction (ASO-PCR) for KIT Asp816Val gene mutation was performed on peripheral blood and proved to be negative.
First consolidation with high-dose cytarabine 3 g/m2 on days 1, 3, and 5 along with idarubicin 12 mg/m2 was given. The course was complicated with pleural effusion recurrence which required therapeutic thoracentesis. Cytology showed no blast cells. Subsequently, the patient received 2 further consolidation courses with high-dose cytarabine that was complicated with febrile neutropenia with Enterobacter cloacae bacteremia on third cycle that required intensive care admission and antimicrobial treatment with meropenem. Bone marrow evaluation after completion of therapy revealed no increase in blast cells, but there was persistence of mastocytosis. As there was a high risk of relapse, allogeneic stem cell transplant was planned. However, it was not done as the patient traveled back to his home country.
Discussion
World Health Organization Classification of Tumors of Hematopoietic and Lymphoid Tissue (2008 edition) recognizes 4 major subcategories for systemic mastocytosis (SM): indolent SM (ISM) with little or no evidence of organ dysfunction, aggressive SM (ASM) with the presence of disease-related organopathy, SM associated with a clonal hematologic non-MC lineage disease (SM-AHN), and mast cell leukemia (MCL) with the presence of ≥20% MC in BM aspirate.1,2
The WHO defines 1 major criterion and 4 minor criteria for the diagnosis of SM. The major criterion is detection of multifocal, dense infiltrates of MC (≥15 MC in aggregates) in an adequate BM biopsy specimen and/or other extracutaneous organ(s). The minor criteria are as follows: (1) greater than 25% of MC (in the BM or other extracutaneous organ biopsy specimens) are spindle shaped or have atypical morphology, or greater than 25% of the MC in the BM aspirate smear are immature or atypical; (2) an activating point mutation at codon 816 of KIT in BM, blood, or other extracutaneous organs is detected; (3) CD2 and/or CD25, in addition to normal MC markers, are expressed on MC in the BM, blood, or other extracutaneous organs; and (4) serum total tryptase levels persistently exceed 20 ng/mL. The presence of 1 major and 1 minor criterion or 3 minor criteria is required for the diagnosis of systemic mastocytosis. The fourth minor criterion involving elevation of serum total tryptase levels is excluded from the diagnostic criteria in cases of SM-AHN. 1
The diagnosis of SM-AHN is established when WHO criteria for SM and a distinct hematologic non-MC lineage disease are met. In the case reported here, the criteria for the diagnosis of AML with inv(16) were fulfilled and the diagnosis of SM was established based on the presence of multifocal, dense infiltrates of MC in BM biopsy (major criterion) and 2 minor criteria, including morphologically abnormal MC that exhibit an aberrant immunophenotype (CD25 expression). Therefore, this patient was diagnosed with SM-AHN of the AML subtype. The serum tryptase level in our case was elevated (38.5 µg/L). The determination of serum tryptase levels is in principle a good diagnostic and differential diagnostic parameter. Elevated serum tryptase levels, however, are not pathognomonic for SM, as elevated levels can also be detected in approximately 40% of patients with AML 9 and in a variable number of cases with myelodysplastic syndrome (MDS). 10
In SM-AHN, the associated clonal hematological non-MC lineage disorder may be diagnosed before, simultaneously with, or after the diagnosis of SM. The diagnosis of SM-AHN in the BM may be difficult and the diagnosis of SM may be missed/masked at the time of diagnosis, mainly due to the tendency of MC to localize within stroma of BM particles and the compact MC infiltrates in the marrow biopsy may be obscured by the associated hematological neoplasm. 11
This case represents a diagnostic challenge as it appeared to be a classical straightforward case of AML with inv(16) without evidence of mastocytosis at the time of initial diagnosis. However, following AML-directed chemotherapy, the presence of multiple perivascular and randomly distributed focal collections of MC positive for tryptase, CD117, and CD25 was unveiled with the reduction in blast cells that established the diagnosis of SM.
Morphologically, the neoplastic MC have been classified into 3 subtypes: metachromatic blast and atypical MC types I and II. 12 Metachromatic blasts demonstrate blast-like nuclear morphology with several metachromatic granules. Atypical MC type I shows elongated cytoplasmic projections (spindle form), oval eccentric nuclei, and hypogranulation, whereas atypical MC type II typically has bilobed or polylobed nuclei. In our case, there was enhanced proliferation predominantly of atypical MC type II with kidney-shaped, bilobed, or multilobed nuclei with cytoplasmic hypogranulation or irregular metachromatic granule distribution, some with fine nuclear chromatin (promastocyte), together with some atypical MC of type I (Figure 4A and B).
Immunohistochemical stains are useful for identifying MC in tissue sections. Tryptase (cytoplasmic), CD117 (membranous and cytoplasmic), and naphthol AS-D chloroacetate esterase are expressed in normal and neoplastic MC. In addition, neoplastic MC show aberrant expression of CD2 and/or CD25. 12 In our case, the abnormal MC expressed CD25 but not CD2. CD25 is considered more sensitive and specific for detection of neoplastic MC by both FCM and immunohistochemistry, whereas CD2 expression is inconsistent and mostly weak or absent. 13 Moreover, it is reported that CD2 expression was observed significantly less often in SM-AHN than in pure SM patients for unclear reason. 7
The most commonly associated hematologic malignancies are AML, MDS, chronic myeloproliferative disorders, and myelodysplastic/myeloproliferative neoplasms. 5 Most of the published cases of SM associated with AML cover the spectrum of the French-American-British classification of AML, with a majority representing either the M2 or M5 subtype. 4
Based on cytogenetic abnormalities, AML with t(8;21)(q22;q22); RUNX1-RUNX1T1 is the most common subtype of SM-AML.11,14 However, case reports on SM with AML with inv(16)(p13.1;1q22); CBF β:MYH11 seem to be extremely rare. On literature review and up to our knowledge, this is the first comprehensive case report of AML with inv(16) presented with concurrent hidden systemic mastocytosis.
The diagnostic workup for systemic mastocytosis includes molecular analysis for KIT D816V mutation. In the patient reported here, the test for Asp816Val c-kit mutation using ASO-PCR performed outside clinical laboratory was negative (sensitivity of the assay, 0.01% mutated alleles). As the ASO-PCR used for this patient was a qualitative PCR with analytic sensitivity of 0.01%, false-negative results caused by low number of neoplastic cells that is below the detection limit cannot be entirely ruled out. KIT encodes for a receptor tyrosine kinase (TK) present on the cell surface of hematopoietic cells as well as other cell types. Activating KIT D816V mutation is found in greater than 80% of cases with mastocytosis. 15
KIT mutations are frequently detectable as well in a group of patients with core-factor–binding leukemias. It is reported in approximately 22% of AML with t(8;21) and in 30% of AML with inv(16) cases. In such cases, exon 17 D816V is the most common mutation found. 16 Detection of KIT mutations in patients with t(8;21) or inv(16)/t(16;16) is indicative of a worse prognosis. 17 Whether patients with AML and KIT D816V mutation have coexisting occult SM is still a controversial issue. A study by Kristensen et al 18 suggested that SM is uncommon in CBF leukemias positive for KIT D816V mutation. Their study examined the frequency of SM in 20 cases of CBF AML; 4 of 13 cases of AML with t(8;21) and 4 of 7 cases of AML with inv(16) examined carried the D816V mutation assessed by real-time PCR of peripheral blood mononuclear cells. None of the 8 positive cases had evidence of SM by immunohistochemistry for CD117 and tryptase. Johnson et al 11 reported SM in 4 (10%) of 40 patients with AML with t(8;21). KIT D816V mutation was identified in 3 of these 4 patients. In keeping with the latter finding, Fritsche-Polanz et al 19 found high frequency of concomitant mastocytosis and reported SM in 7 of 7 D816V-positive non-CBF AML cases.
In patients with SM-AHN, a clonal relationship between the MC and the associated hematologic non-MC lineage disorder (AHN) component has been sought using KIT and other mutations as markers of clonality. Pullarkat et al 20 examined 1 case of SM-AML with t(8;21) using FISH technique and demonstrated that MC also possessed the RUNX1-RUNX1T1 translocation. Moreover, Bae et al 21 demonstrated the presence of D816V KIT mutation using reverse transcription polymerase chain reaction and RUNX1-RUNX1T1 expression by using FISH in both cell lineages in a case of SM-AML with t(8;21). These studies had demonstrated evidence that neoplastic MC and myeloid leukemic blasts are likely to develop from common hematopoietic progenitors. However, Sotlar and colleagues using laser capture microdissection showed that KIT D816V mutations are not only present in MC but are also variably present in the cells of the AHN component. The frequency of KIT D816V mutation, however, is dependent on the type of ANH. These findings challenge the concept that the SM and AHN components in patients with SM-AHN arise uniformly from a precommitted neoplastic progenitor (stem) cell harboring a KIT and suggest that the SM-AHN category proposed is highly heterogeneous. 6
In cases of SM-AHN, the hematologic disorder must be treated, whereas management options for SM include observation alone, symptom management, supportive measures, and cytoreductive therapy for MC debulking in the setting of aggressive, advanced, or treatment-refractory disease. Histamine H1 receptor antagonists and histamine H2 receptor antagonists are used to control mediator-related symptoms. Interferon alfa (IFN-α) as first-line cytoreductive treatment has activity in all SM subtypes and has been shown to improve dermatological, hematologic, gastrointestinal, and systemic symptoms associated with histamine release. Cladribine (2-CdA) is used as a first-line treatment in cases where rapid MC debulking is indicated or in symptomatic patients who are refractory or intolerant to IFN-α.
In recent years, various TK inhibitors have also been used for SM. The rare SM cases that harbor an imatinib-sensitive KIT mutation or those that are KITD816 unmutated may be appropriate candidates for imatinib treatment.22,23 New TK inhibitors, such as midostaurin, target KIT mutants that are reported to act against aggressive systemic mastocytosis and MC leukemia. 24
Allogeneic hemopoietic stem cell transplantation has been reported to induce continuous remission in a subset of patients with aggressive SM and MC leukemia. 25 An open-label, phase 2 study with midostaurin (multikinase inhibitor) targeting KIT D816V showed efficacy in the treatment of patients with advance systemic mastocytosis, including cases of SM-AHN. 26
In our case, and after discussion with a multidisciplinary team, the specific treatment for AML (appropriate for age and performance status of the patient) was given. The hematologic remission was documented by BM examination after the second induction course. Specific treatment for mastocytosis was not necessary as there were no definite symptoms related to MC mediator release. Allogeneic stem cell transplant was planned as there was high risk of relapse.
In conclusion, this report indicates that the diagnosis of SM with associated clonal non-MC neoplasm may be very challenging in some cases. The diagnosis can be easily missed as aggregates of MC may be overlooked at the time of initial diagnosis of AML and that the concomitant diagnosis of SM with AML requires comprehensive morphologic and immunophenotypic evaluation for detection of abnormal MC proliferation. Based on our experience in this case, we recommend routine screening for the presence of hidden mastocytosis in cases of AML at initial diagnosis by including MC tryptase in the immunohistochemistry panel, particularly in cases of CBF AML. Monitoring of larger number of AML patients with SM may help to assess whether patients’ prognosis is truly affected by the presence of concurrent MC disease and whether specific customized treatment may benefit those patients.
Footnotes
Acknowledgements
The authors gratefully acknowledge the contributions of all the staff of the FCM and cytogenetics laboratories at the National Center for Cancer Care and Research in specimen processing and data collection.
Peer review:
Five peer reviewers contributed to the peer review report. Reviewers’ reports totaled 655 words, excluding any confidential comments to the academic editor.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
FH conceived and designed the experiments. FH, AS and DS analyzed the data. FH and AS wrote the first draft of the manuscript and jointly developed the structure and arguments for the paper. DS and HS contributed to the writing of the manuscript. AA, HO, and DS agree with manuscript results and conclusions. AS, FH, DS, HS, HO, and MY made critical revisions and approved the final version. All authors reviewed and approved the final manuscript.