
Abstract
Kasabach-Merritt phenomenon (KMP) is a rare consumptive coagulopathy associated with specific vascular tumors, kaposiform hemangioendothelioma, and tufted angioma. Kasabach-Merritt phenomenon, characterized by profound thrombocytopenia, hypofibrinogenemia, elevated fibrin split products, and rapid tumor growth, can be life-threatening. Severe symptomatic anemia may also be present. With prompt diagnosis and management, KMP can resolve and vascular tumors have been shown to regress. This review highlights the clinical presentation, histopathology, management, and treatment of KMP associated with kaposiform hemangioendothelioma, and less frequently tufted angioma. A classic clinical case is described to illustrate the presentation and our management of a patient with KMP.
Keywords
Introduction
Kasabach-Merritt phenomenon (KMP) is a life-threatening thrombocytopenic coagulopathy associated with rare vascular tumors, such as kaposiform hemangioendothelioma (KHE), and less frequently with tufted angioma (TA). First described by Kasabach and Merritt
1
in 1940, KMP is characterized by severe thrombocytopenia and hypofibrinogenemia. Anemia and elevated
Kaposiform Hemangioendothelioma and Tufted Angioma
Clinical manifestations
Kasabach-Merritt phenomenon is specific to KHE and TA, occurring in 70% of KHE cases 5 and less frequently with TA. Kaposiform hemangioendothelioma and TA are rare vascular tumors, which typically present during infancy or early childhood. Kaposiform hemangioendothelioma most commonly appears as an enlarging, firm, solitary, purpuric cutaneous or soft tissue lesion, although approximately 10% of patients with KHE lack skin involvement. 5 Often multiple tissue planes are involved and tumor borders are ill-defined. 3 Kaposiform hemangioendothelioma typically involves the extremities, trunk, and retroperitoneum.3,6 The risk of KMP is greater with increased depth and infiltration of the vascular tumor and with retroperitoneal or intrathoracic involvement. 5 Tufted angioma similarly occurs on the trunk and presents with violaceous macules and plaques.
Histopathology
Kaposiform hemangioendothelioma and TA arise from capillary and lymphatic endothelium. Kaposiform hemangioendothelioma is an infiltrative tumor, typically involving the dermis, subcutaneous fat, and muscles. Histologically, KHE is marked by irregular sheets of spindle-shaped endothelial cells and characteristic slit-like vascular channels, 7 with positive immunohistochemical staining for lymphatic markers, D2-40, LYVE1, and Prox-1, 8 and negative for GLUT-1, the marker for infantile hemangioma. Immunohistochemical stains are also positive for vascular markers CD31 and CD34. 6 Tufted angioma is characterized by irregularly sized nodules or tufts of capillaries in a “cannonball” pattern.7,9 Similar to KHE, TA shares an identical immunophenotype and stains positive for Prox-1, D2-40, LYVE1, CD31, and CD34. 8
Pathogenesis
The intravascular coagulopathy characteristic of KMP is likely secondary to platelet trapping, given the distinct endothelial architecture of the associated vascular tumors. 6 Platelet trapping has been directly illustrated by positive immunohistochemistry for CD61, a marker of platelets and megakaryocytes, within the vascular lumen. 10 Similarly, studies also demonstrate localized consumption of fibrinogen when radiolabeled fibrinogen is infused in the setting of KMP. 11
Diagnosis
Clinically, KMP is often marked by a rapidly enlarging tumor, which may be painful. A constellation of profound thrombocytopenia and consumptive coagulopathy, including hypofibrinogenemia, characterizes KMP, and therefore, laboratory evaluation is essential for the diagnosis. This includes a complete blood count, fibrinogen,
Management
Supportive care
The management of KHE and TA complicated by KMP includes primarily treating the vascular tumor and providing critical supportive care measures. Despite profound thrombocytopenia, severe bleeding is rare and platelet transfusions should be avoided, except for active bleeding and prior to or during surgery. Platelet transfusions can exacerbate intratumoral bleeding 12 and potentiate platelet trapping, therefore likely causing further platelet activation, rapid tumor growth, 13 and pain. 5 In addition, the half-life of platelets is shorter in KMP.13,14 Cryoprecipitate can be administered for active bleeding, for preparation for surgery, or for fibrinogen less than 100 mg/dL. Cryoprecipitate administration should be avoided in asymptomatic patients or for mildly low fibrinogen values. Packed red blood cells (PRBC) should be transfused for symptomatic anemia only. Heparin is contraindicated in KMP. 3 Adequate pain management is also critical in managing patients with vascular tumors associated with KMP as tumor engorgement by blood elements can cause severe pain and mass effect on the surrounding tissues.
Surgical management, endovascular intervention, and radiation
Complete surgical resection offers the most definitive cure for small, localized tumors; for tumors that have decreased in size with medical therapy; or for life-threatening tumors. 15 However, surgical intervention at initial presentation with KHE is rarely feasible, given the infiltrative nature of the associated tumor and existing coagulopathy.
Use of radiation therapy has been reported in KMP.16,17 A response rate of 75% has been described with the use of radiation in conjunction with steroids 18 ; however, radiation may have long-term complications, including growth arrest, developmental delay, and secondary malignancies, and therefore should be used with caution in younger patients.
Successful use of embolization has also been described in patients with KMP. 19 Potential side effects include risk of skin necrosis secondary to infarction of surrounding tissue and potential for life-threatening bleeding with the use of intraprocedural heparin with catheter placement.
Pharmacologic management
Invasive interventions within the context of coagulopathy are associated with the potential for high-risk complications; therefore, pharmacologic management is often first line and can achieve hemostatic stability in KMP (Table 1). Various small case series using monotherapy or multimodal therapies have been described involving steroids, vincristine,20,21 interferon-alfa,22,23 antiplatelet agents, propranolol,24,25 and sirolimus with variable outcomes 26 and long-term side effects.
Recommended pharmacologic therapies for KHE/TA with KMP.
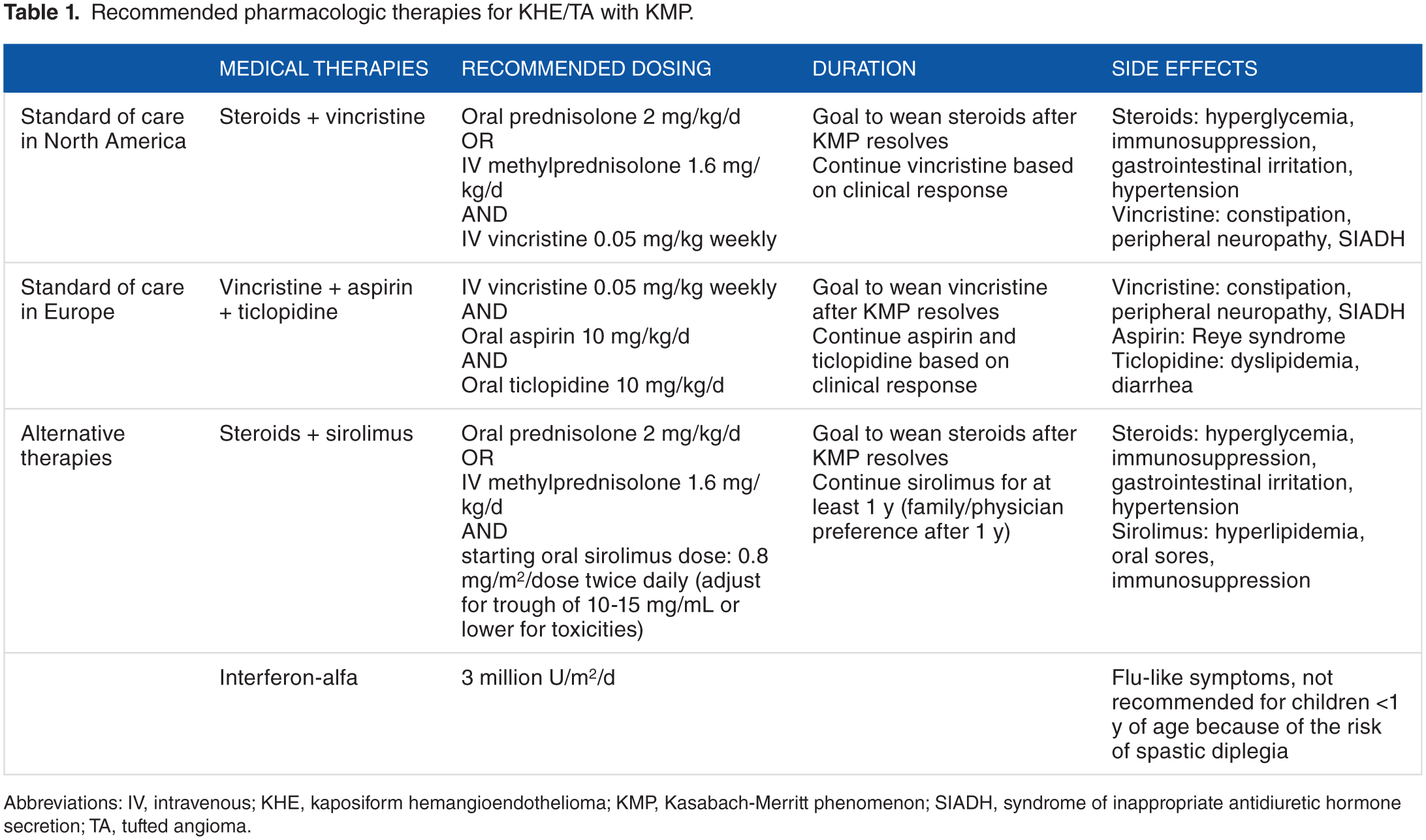
Abbreviations: IV, intravenous; KHE, kaposiform hemangioendothelioma; KMP, Kasabach-Merritt phenomenon; SIADH, syndrome of inappropriate antidiuretic hormone secretion; TA, tufted angioma.
Due to the large variability in management protocols, consensus-derived practice standards have been developed in 2013 by a multidisciplinary expert panel in North America from various institutions to provide a uniform approach to treat KHE with KMP and therefore allow for a cohesive comparison of responses and long-term outcomes. 27 The group recommends a regimen of systemic corticosteroids and weekly vincristine as standard of care for KHE associated with KMP.27,28
During the same time, a phase II study 29 evaluating the safety and efficacy of sirolimus, an inhibitor of mammalian target for rapamycin, for the treatment of complicated vascular anomalies included 10 patients with KHE and KMP, and the response rate was impressive: 100% partial response of KHE, but with complete and quick resolution of KMP in all patients. The number of cases was small (as the incidence of the vascular tumors is low). However, considering the potential side effects of the 2 regimens (vincristine + steroids vs sirolimus + steroids) and mode of administration (intravenous for vincristine vs oral for sirolimus), many centers have adopted sirolimus + steroids as a first-line pharmacologic therapy. A multi-institutional study is on-going to randomly assign patients to the 2 arms described above and evaluate treatment response.
European groups have described successful outcomes with vincristine, aspirin, and ticlopidine in the treatment of vascular tumors with KMP.30,31 Both aspirin and ticlopidine potentiate the inhibition of platelet aggregation. Successful use of aspirin and ticlopidine 32 and vincristine and ticlopidine 33 in KMP has also been described.
Given the various therapies available to manage KMP, a joint decision by the physician and the family should be established regarding the best approach. Although vincristine and steroids have been proven beneficial, this therapeutic approach usually involves placement of a central line in a patient who initially presents with significant coagulopathy. Vincristine is a cytotoxic drug with potential side effects, which include peripheral neuropathy, constipation, and syndrome of inappropriate antidiuretic hormone secretion. Steroids also have side effects, which include hypertension, hyperglycemia, adrenal insufficiency, and gastritis.
The vincristine, aspirin, and ticopidine regimen avoids use of steroids; however, aspirin use in children is associated with the potential risk of Reye syndrome, which is characterized by encephalopathy and liver failure, and therefore should be used with caution in children.
Sirolimus with or without steroids is a therapeutic option that does not require a central line placement as both medical treatments are oral. Blood draws are necessary to assess the sirolimus level and adjust the dose based on the targeted value. These regimens have the potential side effects of steroids, as described above, in addition to the side effects of sirolimus, which include decreased immunity, mucositis, and dyslipidemia.
The length of therapy is determined by resolution of KMP. Improvement in thrombocytopenia can be noted in days after initiation of pharmacologic therapy with significant improvement in coagulopathy in a few weeks. A global consensus on the best first-line therapy is yet to be established.
A Clinical Case: Presentation and Management of Kasabach-Merritt Phenomenon
To illustrate the presentation and management of KMP, we report 1 patient diagnosed at 5 weeks of age with KMP associated with KHE (Figures 1 and 2). This patient initially presented with a soft tissue mass on the nape of the neck and occipital scalp (subcutaneous location). Within a couple of days after presentation, the mass enlarged very rapidly, became tender, and the overlying skin changed from mildly erythematous to a purpuric hue.

Classic clinical case of Kaposiform hemangioendothelioma with Kasabach-Merritt phenomenon (KMP) diagnosed in a 5-week-old patient. Diagnosis, response to blood product transfusion, and resolution of KMP with sirolimus + steroids therapy. (A) Platelet counts (normal: 150-450 × 103/µL), (B) hemoglobin (normal: 9.5-13.5 g/dL), (C) fibrinogen (normal: 220-440 mg/dL), and (D)
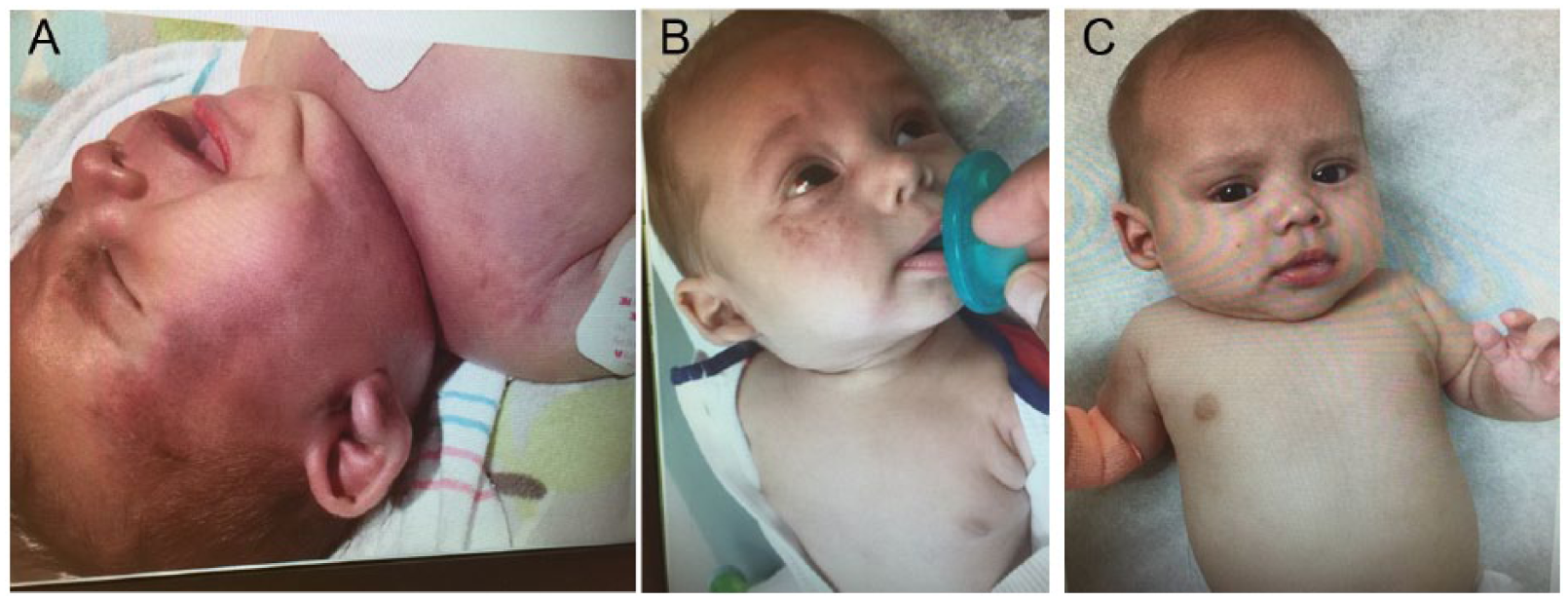
Classic clinical case of Kaposiform hemangioendothelioma with Kasabach-Merritt phenomenon (KMP) diagnosed in a 5-week-old patient. Extension of cutaneous involvement. Photos presented with mother’s permission. (A) At the peak of activity of KMP (day 4)—patient with purpura, edema, irregular margins, and extensive size. (B) After 3 weeks of treatment, the cutaneous component improved significantly. (C) After 2 months of therapy with sirolimus and steroids, complete resolution of the cutaneous component was achieved.
Initial laboratory evaluation of our patient revealed anemia (hemoglobin, 6.8 g/dL), mild thrombocytopenia (platelets, 137 × 103/µL), low fibrinogen (164 mg/dL), and high
Our patient received a single platelet transfusion on day 4 for a platelet count of 2 × 103/µL prior to placement of a central line. The platelet count improved immediately after transfusion (Figure 1) and the patient tolerated the procedure well with no complications. However, within 24 hours of the platelet transfusion, severe thrombocytopenia was again noted with a platelet count of 13 × 103/µL. The patient received a PRBC transfusion on the day of presentation, prior to being diagnosed with KHE and KMP. Hemoglobin improved from 6.8 to 10 g/dL; however, within 4 days, it was again low at 7.4 g/dL (Figure 1). A second PRBC transfusion was administered prior to placement of the central line for hemoglobin of 6.1 g/dL with brief improvement to 9.1 g/dL.
For our patient, we chose sirolimus and steroids as therapy, as per institution preference. Treatment was initiated on day 2 after initial presentation, and a therapeutic trough level for sirolimus was achieved on day 8. The KMP stabilized within a couple of days, and prior to completion of 5 days of therapy, thrombocytopenia, hemoglobin, and fibrinogen significantly improved (Figure 1). Figure 1 illustrates the first month of therapy to demonstrate the classic effect of therapy on KHE and KMP. After 3 weeks of treatment, the cutaneous component improved significantly (Figure 2A) and a steroid wean was initiated. After discontinuation of steroids, the KMP did not recur. Our patient has now completed 4 months of therapy and continues to receive sirolimus monotherapy. The cutaneous component of the patient’s KHE has now resolved as well (Figure 2C).
Conclusions
Prompt diagnosis and initiation of therapy are critical in KMP, which, if not treated, is associated with high mortality. With successful management, KMP can resolve and KHE/TA can regress. A higher mortality may be seen in retroperitoneal lesions, likely secondary to delayed diagnosis and increased association with KMP. Mortality is highly associated with degree of coagulopathy. Long-term complications include recurrence potential in addition to lymphedema, chronic pain, and functional limitations. For our patient with KMP and KHE, we elected to start steroids and sirolimus based on institutional experience and preference. The steroids were weaned after the KMP resolved and the patient continues to tolerate sirolimus with complete resolution of KMP and KHE. Given the rarity of vascular tumors and the associated KMP, large collaborative prospective studies are essential to further identify effective treatment regimens for this life-threatening phenomenon.
Footnotes
Acknowledgements
The authors acknowledge all the members of the Texas Children’s Hospital Vascular Anomalies Center where teamwork and expertise together benefit our patients.
Peer review:
Five peer reviewers contributed to the peer review report. Reviewers’ reports totaled 331 words, excluding any confidential comments to the academic editor.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interest:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
PM and II conceived and designed the experiments, analyzed the data, and jointly developed the structure and arguments for the paper. PM wrote the first draft of the manuscript. PM, JM, II contributed to the writing of the manuscript, agree with manuscript results and conclusions, and made critical revisions and approved the final version: All authors reviewed and approved of the final manuscript.