
Abstract
SAPHO (synovitis, acne, pustulosis, hyperostosis, and osteitis) is a rare syndrome mainly characterized by cutaneous and osteoarticular manifestations. The most typical osteoarticular manifestations are localized to the anterior chest wall and include a usually noninfectious osteitis, hyperostosis, and synovitis of the sternoclavicular joints. However, clinical presentation of SAPHO syndrome can be quite heterogeneous. Several clinical and radiological features are shared with other well-defined pathological entities, and clinical signs and symptoms often occur at different timepoints. Mainly due to this complexity and its rarity, there are currently no validated diagnostic criteria for SAPHO syndrome. Inflammation of the soft tissues around the bones and possible nerve compression could contribute to dysphagia, hypophonia, or obstruction of the airways. Neurologic manifestations could therefore be part of this multiorgan involvement. Here, we present a case of SAPHO syndrome with atypical onset symptoms, characterized by left vocal cord paralysis, acute neck pain due to osteolytic atlantoepistrophic lesion, and an unusual cutaneous manifestation, diagnosed as mid-dermal elastolysis. The latest two, to the best of our knowledge, have been here first described in a case of SAPHO syndrome.
Introduction
The acronym SAPHO was first used by Chamot et al 1 in 1987 to describe a rare syndrome mainly characterized by cutaneous and osteoarticular manifestations, namely, synovitis, acne, pustulosis, hyperostosis, and osteitis. The most typical osteoarticular manifestations include a noninfectious osteitis, hyperostosis, and synovitis of the anterior chest wall, with the sternoclavicular joints commonly affected and with typical radiological findings. 1 Because of the possible involvement of the axial skeleton, the occurrence of enthesitis, and the associations with inflammatory bowel disease, SAPHO had been initially classified as seronegative spondyloarthritis (SpA). 2 However, recent evidence suggests that SAPHO syndrome fits better a primitive inflammatory osteitis, in the spectrum of autoinflammatory diseases (AIDs). 3
The clinical presentation of SAPHO syndrome can be quite heterogeneous. Several clinical and radiological features are shared with other well-defined pathological entities, and clinical signs and symptoms often occur at different timepoints 4 ; therefore, diagnosis may be sometimes difficult, 4 explaining why SAPHO syndrome is often underdiagnosed and considered a rare disease. 5
Mainly due to this complexity and its rarity, there are currently no validated diagnostic criteria for SAPHO syndrome. 6 The most frequently used ones are those suggested by Kahn and Benhamou, requiring pathological evidence of osteitis or osteomyelitis for establishing the diagnosis, with or without typical skin lesions.5-7
Therefore, SAPHO syndrome may represent a real diagnostic challenge. 8 Here, we present a case of SAPHO syndrome with atypical onset symptoms characterized by acute neck pain due to osteolytic atlantoepistrophic lesion and left vocal cord paralysis, associated with an unusual cutaneous manifestation.
Case Description
In May 2018, a 56-year-old white woman was admitted to our Rheumatology Unit for persistent inflammatory pain of the cervical spine, which arouse acutely about 3 months earlier in the absence of traumatic events.
In her medical history, she had suffered from noninflammatory chronic low back pain over the previous 10 years. In 2013, lumbar magnetic resonance imaging (MRI) showed L4-L5 spondylolisthesis with stenosis and osteo-arthritic right facet deformation, successfully treated with local steroid injections and physical therapy.
After admission, neck MRI revealed hyperintensity in T2-weighted sequences on the articular surfaces of the right atlo-epistrophic joint (Figure 1A), with contrast enhancement surrounding a large area of osteolysis (Figure 1B). Anterior somatic and posterior disk osteophyte bars slightly imprinting the medulla anterior profile without impairing the spinal cord were also appreciable (Figure 1C). Computed tomographic (CT) scan confirmed the presence of the osteolytic lesion on the right side of the atloaxial joint with irregular margins due to the presence of fine erosion (Figure 1D and E). A mild posterior subluxation of the atlantoaxial joint with right dislocation of the epistrophic tooth with respect to the atlas was documented, together with the presence of an areola of cortical erosion on the right side of the apex of the tooth (Figure 1F). Although atlantoaxial alterations were first judged compatible with synovitis, a possible neoplastic origin of the osteolytic area could not be ruled out confidently by the radiologist. Consequently, the patient underwent total-body [18F]2-Deoxy-2-fluoro-
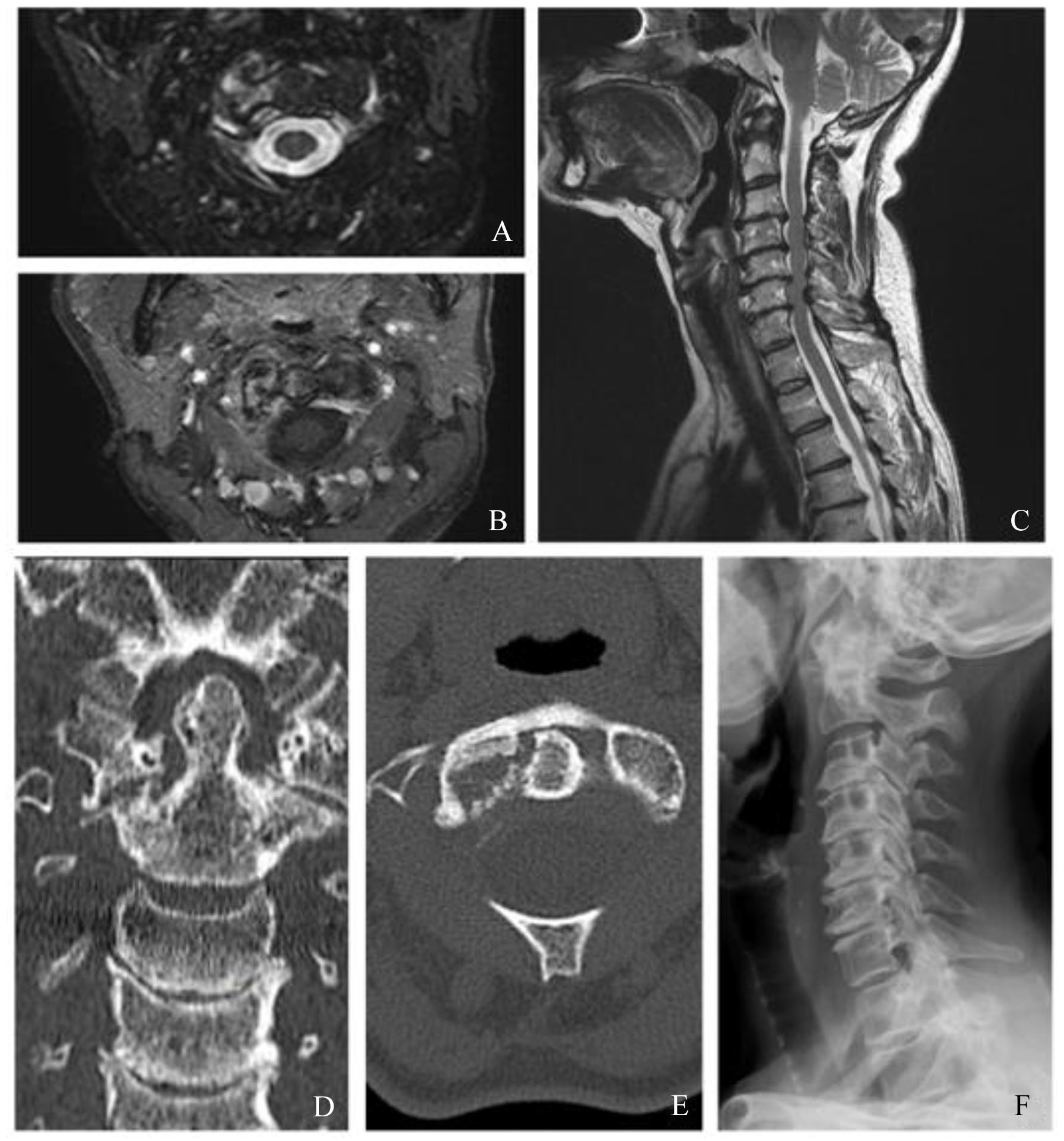
Cervical magnetic resonance image (MRI) (A, B, C) showing areas of altered signal, hyperintense in T2-weighted images involving the articular surfaces of the joint between the atlas and epistrophe on the right side (A: axial fat suppression T2-weighted sequences), gaining the contrast medium and surrounding a large area of osteorarefaction compatible with osteolysis (B: axial postcontrast T1-weighted images). Voluminous anterior somatic osteophytes (not significantly compressing the esophagus) and posterior osteophytic disk bars which mark the anterior profile of the medulla at several levels (C: sagittal fat suppression T2-weighed sequences). No areas of altered spinal cord signal are detected. Neck computed tomographic (CT) scan (D, E). As partially visible in neck MRI, in CT images the focal osteolytic alteration involving the right side of the apex of the tooth and the atloaxial joint, associated with irregular bone margins due to the presence of fine erosions, is confirmed (D: coronal view; E: axial view). (F) shows lateral radiography of the cervical spine, which demonstrated a posterior dislocation of the atlas with respect to the axis. Simultaneously, aspects of spondyloarthrosis are appreciable in the C3-C7 cervical tract with reduction in thickness of almost all the related intersomatic spaces.
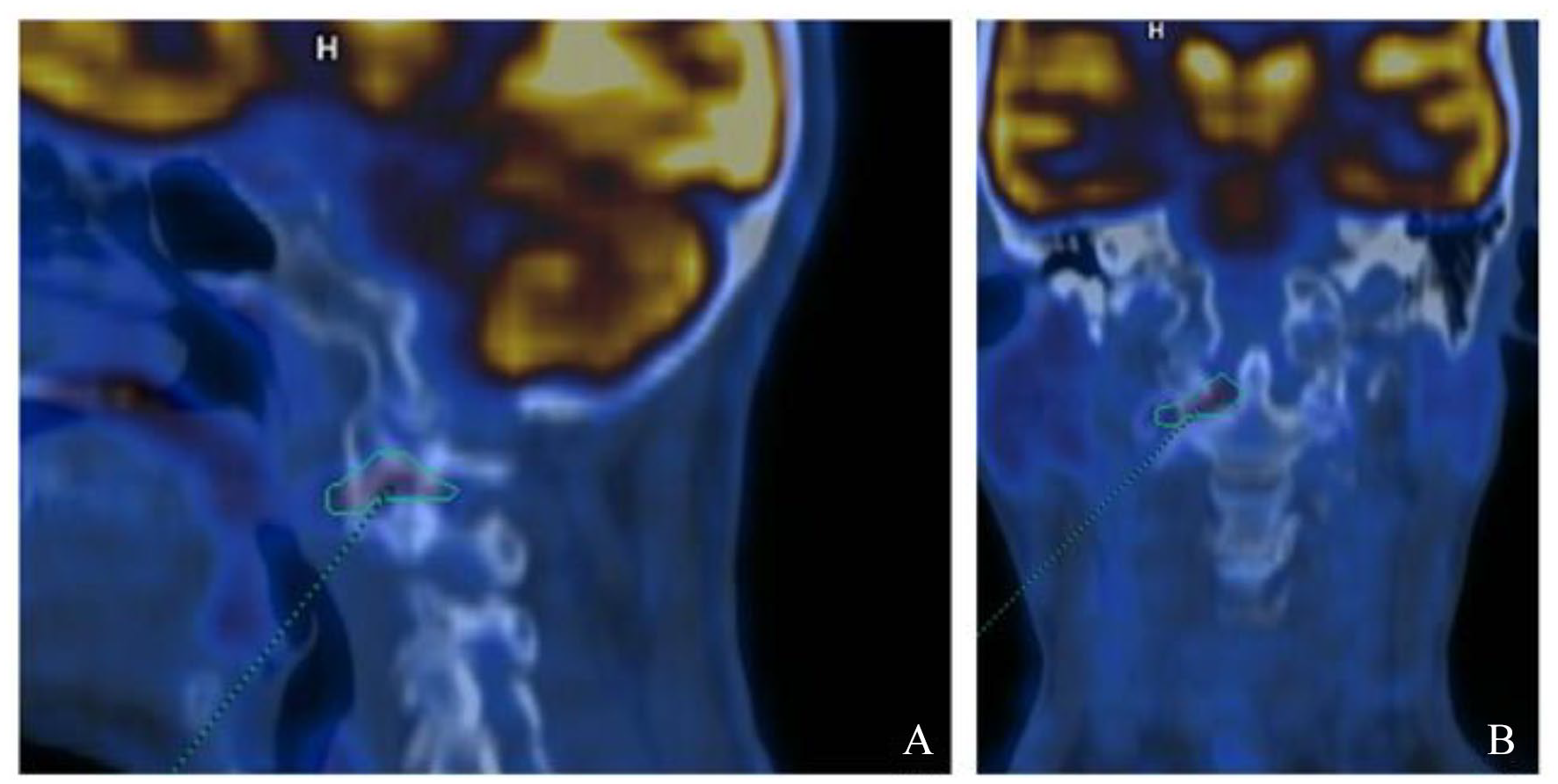
Whole-body [18F]2-Deoxy-2-fluoro-

Bone scintigraphy exhibiting modest hyperfixation in correspondence with the articular passage between the atlas and the epistrophe of the right side, site of known osteo-structural alterations described in magnetic resonance imaging and computed tomographic images. Mild hyperfixation at the scapular girdle, lumbar L4-L5 levels (site of known spondylolisthesis), and trapeziometacarpal joint of both hands, on a degenerative basis.
On February 27, 2019, the patient was admitted to the local Emergency Department with a 10-day history of severe and progressively worsening pharyngodynia and hypophonia, neck lymphadenopathy, intense pain in the right mastoid-occipital region, severe dysphagia for both liquids and solids, and diffuse erythematous-wheal patches on the thighs. Blood pressure on admittance was 140/100 mm Hg and heart rate was 100 bpm. Laboratory findings showed slight increase in inflammatory markers (ESR: 43 mm/h; CRP: 1.5 mg/dL) and mild neutrophilic leukocytosis (white blood cell [WBC]: 13 500 cells/µL, cutoff <11 000 ESR: 43 mm; neutrophils 8100 cells/µL, cutoff <7700 cells/µL), with normal liver and kidney function.
An ear-nose-throat (ENT) consultation and laryngoscopic examinations revealed paralysis of the left vocal cord in paramedian position as well as salivary stagnation in left retrocricoid region and piriform sinus and tremors of the left arytenoids; good compensation in adduction by the contralateral vocal cord was appreciated, and no lesions in the mucosa of oral cavity, nor hypopharynx or larynx were detected. A clear right velar motility deficit was also recognized. A CT scan of the neck, mediastinum, and chest excluded the presence of compressive masses on IX and X cranial nerve and in particular on the left recurrent laryngeal nerve. Brain CT scan and esophagogastroduodenoscopy did prove also normal. Ultrasound (US) examination of the neck bilaterally revealed some enlarged lymph nodes with reactive features; localized abscess, expansive lesions, or pathological findings of the submandibular glands were excluded. No fever, dyspnea, chest pain, or other systemic symptoms were detected. Steroid therapy with methylprednisolone 80 mg/day was then scheduled, and at the beginning of March 2019, she was transferred to the Hospital Unit of Clinical Neurology. On admittance, neurological examination revealed severe hypophonia with slightly bitonal voice, slight reduction of left eyelid, lower left palate but mildly hyporesponsive bilaterally, and diffuse hyperreflexia in 4 limbs. No deviations of the tongue in the oral cavity or during protrusion were observed. The patient also reported pain and swelling sensation in the right occipital-nuchal region with painful paresthesias in the territory of the right great occipital nerve, especially triggered by eversion movements of the head on the right and with irradiation to the right hemitongue. The right ipsilateral upper arm showed slight diffuse swelling.
A number of clinical, laboratory, and instrumental examinations were undertaken mainly aimed to rule out bulbar involvement from either a disorder of the neuromuscular junction or a cranial multifocal neuropathy.
The search for anti–acetylcholine receptor (AchR), anti-muscle-specific kinase (MUSK), anti-ryanodine receptors, anti-titin, and anti-ganglioside antibodies was negative. An extensive infectious panel, including blood cultures, viral hepatitis workup, parvovirus B19 and EBV serology, and HIV screening, was negative. Single-fiber electromyography (SFEMG) of the frontal muscles showed mild abnormalities of the neuromuscular junction.
Brain MRI proved negative. Alterations of the skull bones, in particular hyperostosis or osteomyelitis, as well as dural thickening, were excluded. The MRI of the cervical spine cord did not show significant changes compared with that performed in May 2018, and no masses or lesions potentially affecting IX and X cranial nerves were detected. At lumbar puncture, hyperproteinorrachia (140 mg/dL) and minimal WBC content (5/µL) were found. Electrophysiology of the 4 limbs was normal. Cerebrospinal fluid culture was negative. The US of supra-aortic, vertebral, and subclavian arteries found no abnormalities, while at the level of the neck the lymphadenopathy was no longer appreciable. A venous Doppler ultrasound of the right upper limb and neck ruled out venous thrombotic lesions and showed diffuse mild subcutaneous lymphedema. Patent foramen ovale was excluded through transthoracic echocardiography and transcranial Doppler.
After rheumatological consultation, both primary and secondary vasculitis of the central nervous system and chronic inflammatory autoimmune diseases were ruled out based on clinical, laboratory, and imaging data. Autoimmunity tests such as antinuclear antibody (ANA), antineutrophil cytoplasmic antibodies (ANCA), C3, C4, lupus anticoagulant (LAC), anti-cardiolipin, and anti-beta-2 glycoprotein antibodies and rheumatoid factor were collected and found to be negative. No clinical signs of arthritis or enthesitis in either the right upper limb or other peripheral sites were detected. Power-Doppler ultrasound of shoulder and elbow excluded subclinical synovitis, tenosynovitis, or enthesitis.
Dermatologist consultant highlighted localized scattered papules and erythematous patches in the thighs and programmed a skin biopsy.
Based on the reported clinical, laboratory, and imaging data, disorders of the neuromuscular junction could be ruled out and a diagnosis of cranial multifocal immune-mediated neuropathy (IX and X nerves) was considered the most likely. High dose of intravenous methylprednisolone (500 mg/day for 7 days) was started with a prompt, although moderate, response, followed by gradual tapering with oral prednisone for the next 45 days. Meanwhile, the histopathological result of the skin biopsy was available, leading to a diagnosis of mid-dermal elastolysis (Figure 4)
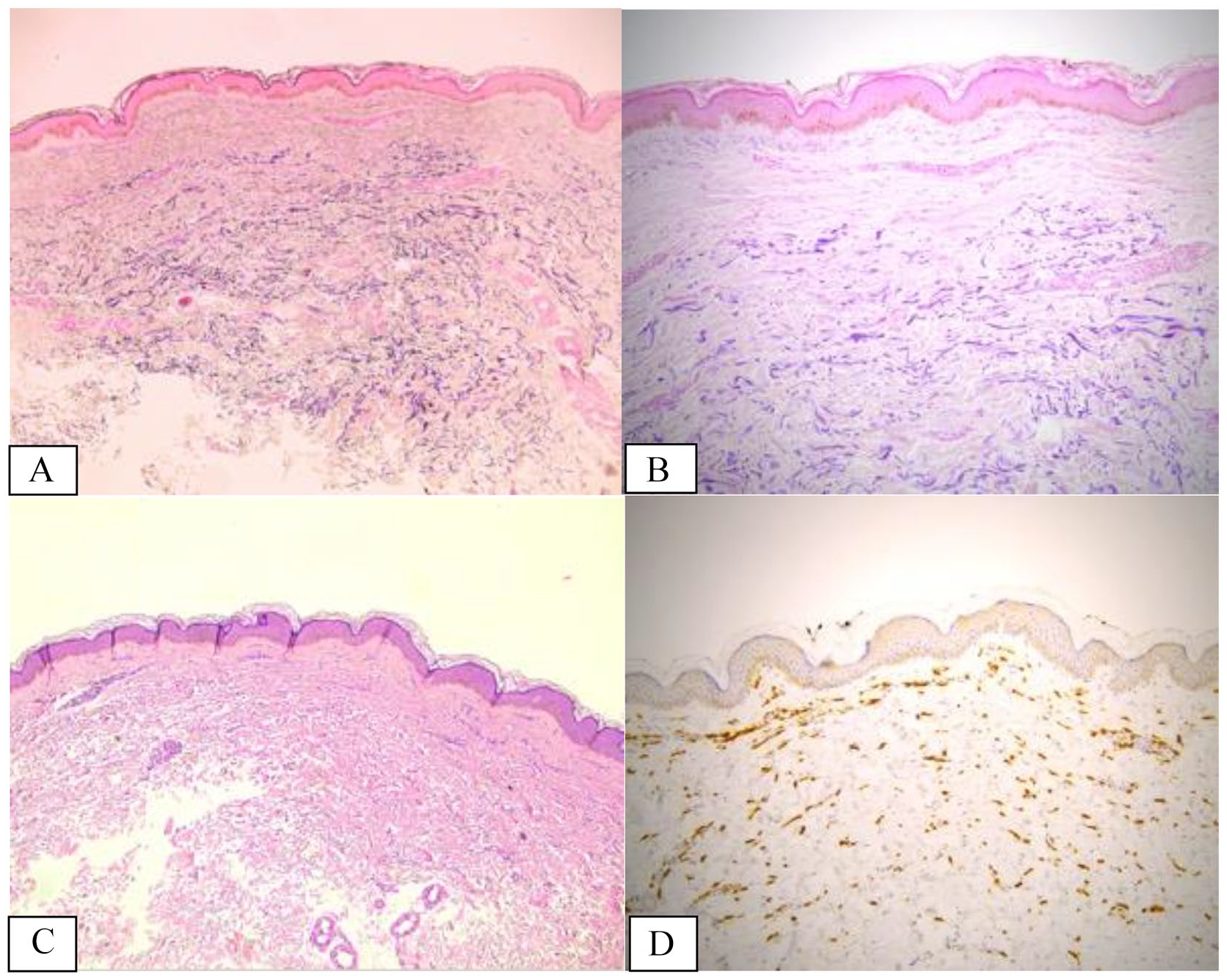
(A and B) Histopathological finding revealing the band-like complete loss of elastic fibers in the mid-dermis, annotated with dotted lines, properly studied by elastic stains; elastic fiber fragmentation is also detected (Weigert stain, 5× magnification/A and 20× magnification/B). (C) Hematoxylin and eosin staining did not reveal pathological findings except for slight lymphohistiocytic perivascular infiltrates around the vessels of the superficial plexus in the lesional sites. There is no evidence of dermal actinic damage such as elastosis (5× magnification). (D) Immunolabelling for CD163 (a marker of monocytic/histiocytic lineage) revealed a discrete increased number of interstitial histiocytes/macrophages engulfing elastic fibers within the mid-dermis and occasional figures of elastophagocytosis (5× magnification).
On discharge from the neurological unit (March 29, 2019), slowly remitting mild dysphonia and dysphagia for solid food, and slightly hyporesponsive soft palate were detected on clinical examination, and the patient complained of persistent painful paresthesias in the right occipital region. The ENT reassessment documented remission of the velar motility deficit, with left vocal cord remaining in the paramedian position with signs of arytenoid motility and good contralateral compensation. No salivary stagnation in the piriform sinuses was documented. Remission of right upper arm lymphedema was recorded.
The patient remained clinically stable until September 2019 when severe arthralgias in hands, knees, and feet; inflammatory low back pain; and painful swelling in the right sternoclavicular region appeared. The patient was then readmitted to the Rheumatology Unit. Radiograph of the dorsal and lumbar spine, pelvis, knees, hands, and feet showed diffuse mild degenerative alterations of the cartilage without signs of inflammatory involvement, neither erosions nor calcifications. Chest MRI revealed intra-articular effusion with marked bone edema of the medial extremity of the right sternoclavicular joint and millimetric pseudocystic aspects of the bone (Figure 5). Pelvis MRI excluded sacroiliitis. The US of the hands showed the presence of active subclinical synovitis of II and III left metacarpophalangeal (MCP) and II-IV right MCP joints, while active enthesitis of the insertion of extensor tendons was appreciated on the elbow.
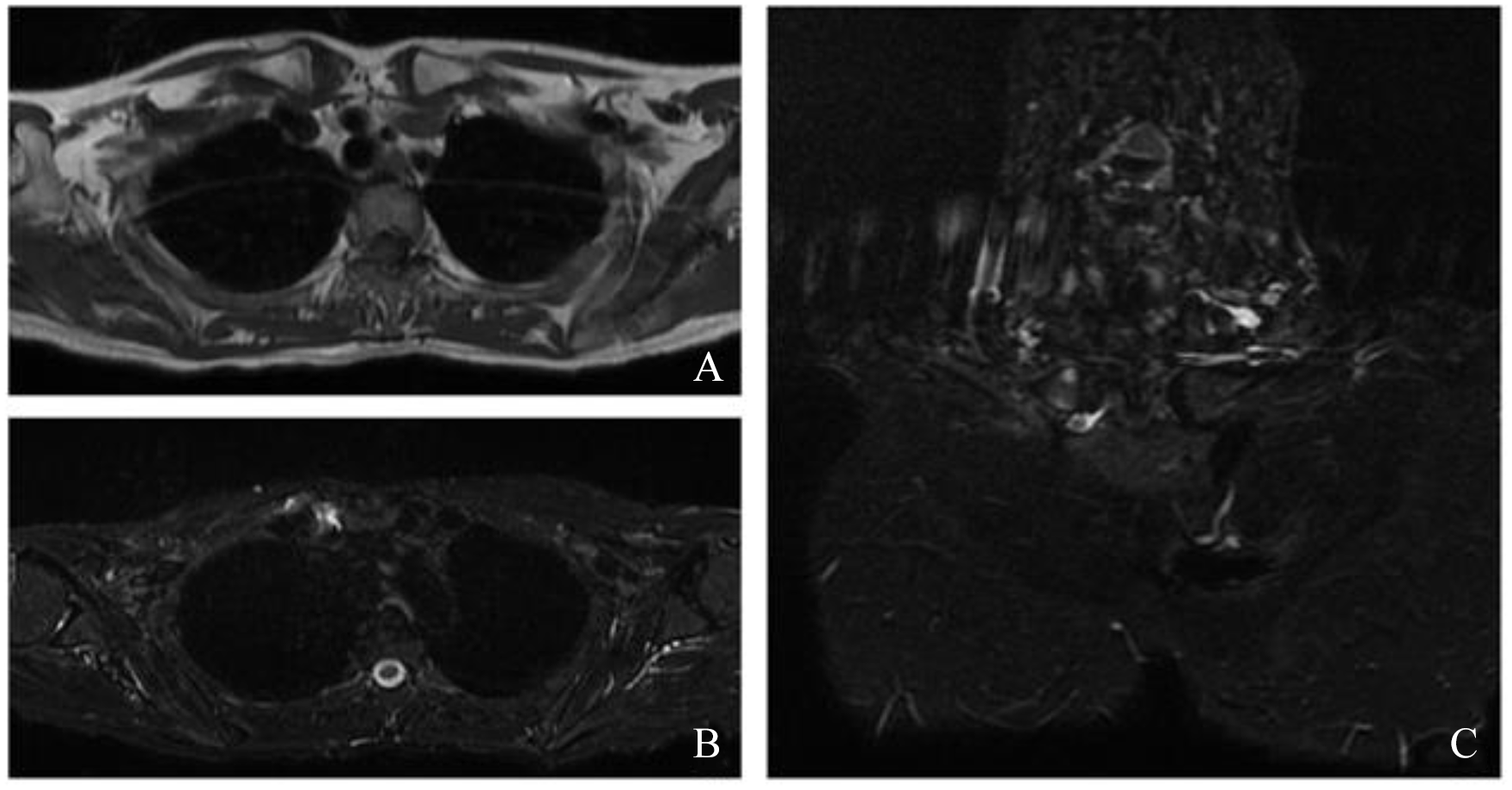
Chest magnetic resonance imaging. At the level of the right sternoclavicular joint, a periarticular and intra-articular effusion is observed (A: axial T1-weighed image; B: axial Short tau inversion recovery [STIR] image). Reactive sponge edema of the medial extremity of the right clavicle is appreciated, in the context of millimetric areas of hyperintense signal in the long TR sequences in the subchondral area, referable in first hypothesis to pseudocystic aspects or erosions (C: coronal STIR image).
According to both criteria by Kahn and Benhamou,5,7 diagnosis of “SAPHO” syndrome associated with cranial multifocal immune-mediated neuropathy was made.
Therapy with low-dose steroids (methylprednisolone 4 mg/day), methotrexate (MTX; 15 mg/week), and bisphosphonates was then started.
A slight dysphonia and parasthesia at the nuchal level still persisted, even if rather improved, with a persistence of moderate occipital headache, the latter sufficiently controlled by analgesic therapy. Dysphagia and skin manifestations completely remitted. Osteoarticular manifestations were sufficiently controlled by MTX therapy, although periodic phases of painful exacerbation appeared, easily controlled with transient increase in steroid dose.
The ENT reassessments were performed again and substantially overlapped with those obtained in March 2019. Further neurological examinations were normal, and the patient only complained of mildly worsened paresthesias in the territory of the right great occipital nerve, partially responsive to ibuprofen and diclofenac, but not to gabapentin.
In the next 2 years, the patient underwent regular ENT, neurological, and rheumatological follow-up: no other suspicious symptoms occurred, and, in particular, no signs of degenerative neurological disease occurred.
Discussion
SAPHO syndrome is a rare disease, often misdiagnosed and underestimated because of the different and heterogeneous combination of osteoarticular and cutaneous manifestations, frequently occurring at different times, sometimes with a long period of latency between one and another,4,8 therefore representing a diagnostic challenge. 9
Our case represents an example of how the presence of unusual and atypical clinical manifestations, especially if they occur in an asynchronous temporal sequence, can lead to significant diagnostic pitfalls.
The first symptom that brought the patient to our attention was inflammatory acute neck pain resulting from inflammatory involvement of the atlantoaxial joint. The upper cervical structures (C1 and C2 vertebrae, including the atlanto-axial, atlanto-odontoid and atlanto-occipital joints) can be involved in several musculoskeletal inflammatory diseases, such as RA, SpA, crystalassociated arthropathies (especially chondrocalcinosis), and septic arthritis, but also noninflammatory conditions such as diffuse idiopathic skeletal hyperostosis (DISH), 10 all excluded based on clinical and radiological data.
Nevertheless, given the uncertain and suspicious nature of the osteolytic lesion, the patient also underwent a FDG-PET investigation, which excluded the presence of neoplasms as well as any inflammatory involvement of peripheral joints, revealing only mild hypercaptation at the atlo-epistrophic level. Bone scintigraphy showed mild hypercaptation at the same level, and both results were judged compatible with active synovitis of the atlantoaxial joint.
In SAPHO syndrome, cervical spine as well as dorso-lumbar spine and sacroiliac joints may be involved with several aspects similar to those seen in SpA.11,12 In particular, paravertebral ossification with nonmarginal and asymmetric new bone formation resembling PsA can be observed, leading to bony bridging across the disco-vertebral junction and evolving toward vertebral fusions. 13 Again, in our case, the cervical lesions appreciable with MRI and CT images were typical osteophytes due to osteoarthritis (OA). To the best of our knowledge, there are no previously described cases of inflammatory atlanto-axial involvement in SAPHO syndrome.
The second unusual aspect of our case is the subsequent appearance of severe pharyngodinia, dysphagia, and hypophonia. Among musculoskeletal diseases, these symptoms may be observed in some conditions characterized by exuberant bony proliferation such as DISH, ankylosing spondylitis, or OA, in which vertebral hyperostosis may be so vigorous that pharyngo-esophageal and laryngo-tracheal compression can occur.14-17 Inflammation of the soft tissues around the bones and possible nerve compression could contribute to dysphagia, hypophonia, or obstruction of the airways.18,19 However, in our case, both MRI and CT excluded all these features, and after an in-depth neurological diagnostic workup, a diagnosis of primary inflammatory cranial multineuropathy was made with the main involvement of IX and X cranial nerves. The painful and paresthetic symptoms described in the right occipital-nuchal area were instead considered a consequence of the involvement of the right great occipital nerve at the level of atlanto-axial joint.
In SAPHO syndrome, neurological manifestations have been reported, resulting from compression of vascular or nervous structures by expansive bone lesions and/or osteitis-induced inflammation of the dura mater, and others such as thoracic outlet syndrome, 20 cervical spinal cord injury, 21 sudden deafness or mixed-type hearing loss,22,23 headache, 24 hypertrophic pachymeningitis, 25 and aseptic meningitis with lower cranial nerve palsies. 26 However, primary neurological involvement of central and peripheral nervous system is very unusual and limited to few described cases.
In 2002, Vanin et al 27 first described a case of SAPHO syndrome with an unusual presentation of right acute transitory hemiparesis in an 8-year-old boy. Cerebral CT scan and MRI were both negative, hemiparesis resolved within 2 weeks, and after exclusion of other causes the neurologic manifestation was classified as a reversible ischemic neurologic deficit (RIND). This condition is very rare in children and seems to be the consequence of cerebrovascular lesions, which may derive from different causes,27,28 such as systemic or vascular diseases, chemotherapy, or parenteral nutrition, 29 all conditions excluded in the reported case; for this reason, a possible link between RIND and SAPHO syndrome was hypothesized by the authors. 27
Abul-Kasim et al 30 first reported, in a 41-year-old woman, a case of central nervous system (CNS) involvement in a patient with SAPHO syndrome. Neurological symptoms acutely appeared at the age of 39 years, 8 years after the diagnosis of SAPHO, and they were characterized by numbness of left arm followed by a grand mall epileptic seizure. The MRI evidence on FLAIR sequences showed a lesion at the junction between the cortex and the subcortical white matter of the right parietal lobe. After surgical biopsy, histopathology showed sterile inflammatory changes in the brain parenchyma and the adjacent meninges, which has been considered to be related to SAPHO syndrome. About 1 year after surgery, the patient had a neurological relapse, characterized by right-sided headache and, again, numbness of the left arm, associated with numbness of legs. The MRI showed again a small lesion surrounded by perifocal edema. Interestingly, because clinical symptoms of multifocal osteomyelitis in the extremities appeared at the same time, the patient was treated with biphosphonate infusion (zolendronic acid) and doxycycline, showing not only a marked clinical improvement of the osteoarticular symptoms but also a progressive remission of brain lesions as documented by MRI. 30
Matsuzono et al 31 first reported a primary involvement of peripheral nervous system (PNS) overlapping with SAPHO syndrome in a 53-year-old woman with refractory headache and polyneuritis (I, II, and VII cranial nerves) which occurred before the appearance of skin and osteitis lesions. Our case shares many similarities with that of Matsuzono, because primary inflammatory polyneuritis involving IX or X cranial nerves was diagnosed before the appearance of osteoarticular symptoms.
To the best of our knowledge, there are no other cases of cranial multifocal neuropathy associated with SAPHO syndrome reported in the literature, to date.
About the pathogenesis of this unusual association, it could be speculated that bloodstream spreading of low virulence microorganisms, reported to have an etiological role in SAPHO syndrome or even an immune reactivity to such pathogens, could probably have led to an involvement of both CNS and PNS through the induction of a small-vessel vasculitis-like process.27,30,32
Indeed, in our patient, brain MRI revealed multiple silent punctate lesions predominantly localized in the subcortical white matter of both cerebral hemispheres for which the suspicion of vasculitic or a systemic autoimmune nature was initially raised and ruled out after multidisciplinary consultation; anyway, lacking any histopathological information, they remained of uncertain interpretation.
With the subsequent appearance of sternoclavicular involvement with typical radiological aspects, diagnosis of SAPHO syndrome was established according to both criteria proposed by Benhamou and Kahn. 5 Our patient did not have typical cutaneous involvement, but, as known, SAPHO syndrome can occur without any skin manifestation.8,9,33 In addition, in our patient, almost simultaneously to neurological manifestations, atypical cutaneous lesions appeared in the thighs, which were consistent with mid-dermal elastolysis (MDE) on skin biopsy. Mid-dermal elastolysis is a rare dermatosis with a characteristic mid-dermal loss of elastic tissue on histopathology, whose etiology is still unknown.34-36 However, autoimmune phenomena (eg, elevated ANAs, circulating immunocomplexes) and autoimmune diseases (eg, Hashimoto thyroiditis, Grave disease, lupus erythematosus, RA) have been described in patients with MDE, and an autoimmune pathogenesis has been recently suggested. 37
To the best of our knowledge, no cases of association of MDE with SAPHO syndrome have been described to date. Interestingly, in the subsequent course, our patient also developed autoimmune thyroiditis.
Our case clearly demonstrates not only the diagnostic difficulties but also the therapeutic ones, which can be encountered in treating such a complex syndrome, especially when it is characterized by unusual presentations besides skin and osteoarticular systems, such as nervous system involvement. High-dose steroid therapy was initially required for both the treatment of atlantoaxial synovitis and neurological manifestations. Therefore, after a definitive diagnosis of SAPHO, the patient started immunosuppressive treatment with MTX in combination with bisphosphonates and low doses of steroids. Nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, antibiotics, biphosphonates, and conventional disease-modifying anti-rheumatic drugs (DMARDs), such as MTX, have variable degrees of efficacy over time.8,9 In the last 15 years, the use of biologic drugs has given novel therapeutic insights, and to date, the use of anti–tumor necrosis factor (TNF) agents has proved to be an effective treatment for unresponsive or refractory SAPHO cases.8,38 However, in our patient, the involvement of the nervous system prevented its use. In fact, serious side effects associated with immune suppression have been reported, including central and peripheral nervous system demyelinating disorders. 39 Moreover, the exposure to TNF inhibitors in patients with autoimmune diseases appeared to be associated with increased risk of inflammatory CNS events. 40 Other biologics have been used in SAPHO such as anakinra and more recently ustekinumab and secukinumab, but due to a satisfactory disease control with MTX, they have not been used in our patient.
In conclusion, SAPHO syndrome may represent a real diagnostic challenge especially when different combinations of osteoarticular and cutaneous manifestations occur at different times or when unusual manifestations precede the more typical alterations of the syndrome.
Association of SAPHO with the inflammatory bowel diseases and with less typical skin manifestations such as pyoderma gangrenosum or Sweet syndrome5,8 suggests that this condition may represents a systemic disease. Neurologic manifestations could therefore be part of this multiorgan involvement, as well as synovitis of the atlantoaxial joint and rare skin involvement, such as mid-dermal elastolysis. The latest two, to the best of our knowledge, have been here first described in a case of SAPHO syndrome.
More observations will be needed to confirm whether such atypical and unusual presentation should be considered as part of the wide heterogeneous landscape of SAPHO syndrome manifestations.

Footnotes
Acknowledgements
Not applicable.