
Abstract
One of the most exciting recent hypotheses in neurology is that most neurodegenerative diseases are caused by the neuron to neuron propagation of prion-like misfolded proteins. In Parkinson disease, the theory initially emerged from postmortem studies demonstrating a caudal-rostral progression of pathology from lower brainstem to neocortex. Later, animal studies showed that the hallmark protein of PD, α-synuclein, exhibited all the characteristics of a prion. Here, we describe our work using human neuroimaging to test the theory that PD pathology advances via a propagating process along the connectome. We found that the pattern and progression of brain atrophy follow neuronal connectivity, correlate with clinical features, and identify an epicenter in the brainstem.
The proposal that Parkinson disease (PD) is a propagating neuropathological process was first made by Braak et al. 1 They based this theory on postmortem evidence showing that α-synuclein (Lewy) pathology appears to have a stereotyped distribution, which they classified into 6 stages along a caudal to rostral gradient. Stages 1 and 2 involved the lower brainstem and were seen in asymptomatic individuals who had not shown signs of PD during life. Stage 3 involved the substantia nigra and was associated with symptomatic PD. This is consistent with the fact that the motor symptoms that lead to a diagnosis of PD are due to loss of dopamine neurons. The later Braak stages (4-6) involve the supratentorial central nervous system (CNS), including limbic and neocortical regions, and are associated with cognitive impairment. This distribution of pathology appeared to reflect known neuronal connectivity pathways, leading Braak to propose that a toxic agent originating in the gut and/or olfactory system accessed the CNS via the olfactory and vagus nerves. 2 The gut origin of PD was consistent with several observations: the early involvement of the dorsal nucleus of the vagus, the existence of constipation as an early symptom, the association with pesticides (which could be ingested or inhaled), and the strong links between gut and olfactory signaling and the reward system, which is centered around dopamine neurons. A major role of dopamine in all animals is the signaling and learning of olfactory and food stimuli related to energy homeostasis. Neuronal connections between gut, olfactory system, and dopamine neurons could explain why the disease affects dopamine neurons at an early stage.
More support for the propagating hypothesis came from several postmortem studies in patients who had undergone fetal cell transplantation, a once-promising treatment for PD. Lewy body pathology was observed in the grafted neurons. 3 Moreover, there was evidence that the degree of Lewy pathology was proportional to the time since the grafting procedure. Because the grafts came from embryos that presumably did not have PD, the likeliest explanation was that misfolded α-synuclein could spread from the patient’s affected neurons to the grafted neurons. This mode of neuron to neuron transfer was subsequently proven in a series of seminal animal experiments. 4 For a more thorough historical account of the successive discoveries and slow paradigm shifts that led to the protein propagation model in PD, see the work by Brundin et al (2016). 5
In summary, PD appears to be caused by a normally expressed protein adopting a misfolded state that renders it self-propagating, infectious, and neurotoxic. This had led some researchers to label PD and other neurodegenerative diseases as “prion-like.” Some researchers even go further and simply refer to the abnormal proteins in PD, Alzheimer disease, and other neurodegenerative diseases as prions. 6 It should be noted that the model remains controversial as noted in a recent point-counterpoint debate.7,8 The counterarguments against the prion hypothesis of PD include the following. (1) Known human connectivity does not fully explain the spread of the disease. The substantia nigra is a major node of the disease but its main afferent and efferent projection sites in the basal ganglia appear to be unaffected by Lewy pathology (we address this in our work below). (2) Not all neurons are affected. However, just as infectious epidemics mostly affect vulnerable individuals (eg, the young and elderly), variations in neuronal or regional vulnerability do not argue against a propagating mechanism. Similarly, herpes simplex encephalitis, the prototypical propagating CNS infection, does not kill every neuron. There is selective vulnerability to the spreading infection. 9 Indeed, there is good evidence that neurons have differential vulnerability to α-synuclein–induced neurodegeneration in PD models. For example, variations in α-synuclein protein expression may lead to differences in vulnerability, 10 a fact that further lends support to the prion hypothesis.
Our work seeks to test the propagation model in humans. For this, we have made use of a superb resource: the Parkinson Progression Markers Initiative (PPMI) available at www.ppmi-info.org. 11 The PPMI is a large well-characterized open access database consisting of extensive clinical and imaging data in more than 500 patients with PD and controls. It is a public-private partnership funded by the Michael J. Fox Foundation for Parkinson’s Research and funding partners, found at www.ppmi-info.org/fundingpartners.
The patients with PD in PPMI were originally enrolled soon after diagnosis (<7 months) and prior to starting anti-parkinsonian medications. They underwent T1-weighted magnetic resonance imaging (MRI) (at 1.5 and 3.0 T), single-photon emission computed imaging for assessing striatal dopamine reuptake transporter levels (a measure of dopamine denervation) and clinical and neuropsychiatric assessment. To that point, MRI studies in de novo patients with PD had tended to show very little to no brain atrophy. However, we leveraged the large sample size (237 patients with PD and 118 age-matched controls scanned at 3.0 T) and 2 image analysis techniques: deformation-based morphometry (DBM) and independent component analysis (ICA), to identify a PD-specific pattern of brain atrophy. 12 Deformation-based morphometry uses information from the nonlinear transformation of each individual brain to a template to generate maps of local tissue expansion or retraction. We provided evidence in the replies to reviewers of this article (available at https://elifesciences.org/articles/08440#author-response) that DBM is especially suited to detecting subcortical tissue atrophy.
We found that the pattern of brain atrophy in these patients with early PD nicely recapitulated the schema proposed by Braak et al (Figure 1). But how can one prove a propagating process based on data from a single time point? As is the case for infectious diseases propagating through a population, it is possible to infer a spreading process and an epicenter from the disease distribution at a single time point, 13 as long as one knows the route of propagation. This is how John Snow, the founder of modern epidemiology, traced a London cholera epidemic to the Broad Street water pump (https://en.wikipedia.org/wiki/1854_Broad_Street_cholera_outbreak).
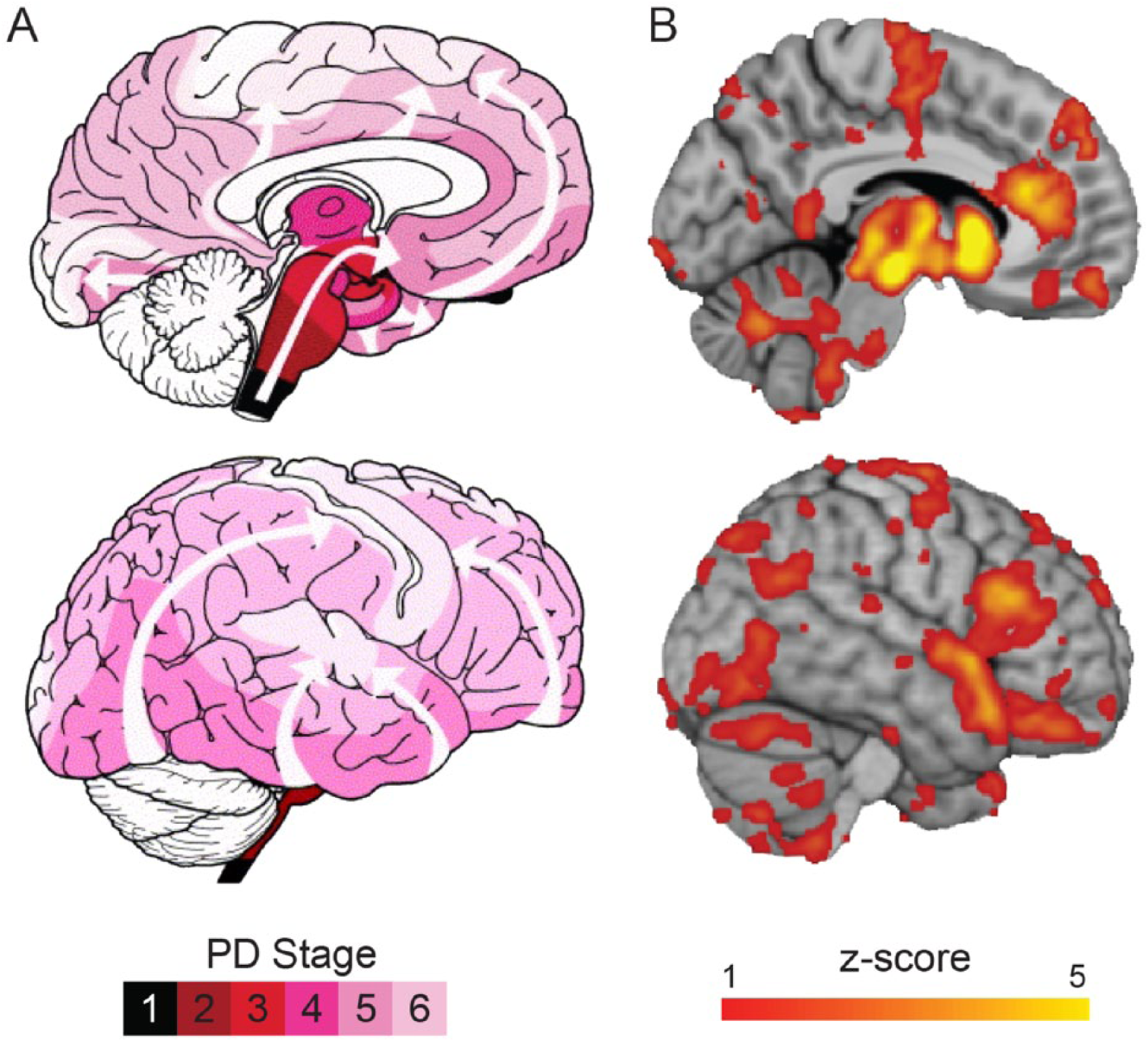
We were also able to show that the pattern of atrophy fit with propagation via the human connectome—the white matter network that connects all cortical and subcortical brain regions and that the likeliest epicenter was the substantia nigra. 12
Another aspect of propagating diseases is that the spatial pattern of disease spread will be constrained by the potential routes of spread. In the case of neurodegeneration, the expression of the disease is expected to be determined by the connectome. This is a suggestion that was first tested in Alzheimer disease and other dementias.14,15 We used partial least squares (PLS) to look for patterns of covariance between brain atrophy and clinical features in the PPMI cohort and identified 3 spatial modes of disease spread associated with a typical, a severe, and a milder progression. 16 The “typical” progression has a spatial pattern that is also observed in normal aging and corresponds to the Braak staging scheme. The more severe expression appeared to involve earlier propagation of the disease to limbic and prefrontal areas, possibly accounting for dementia, mood disturbances, and hallucinations, seen in more severe/advanced cases of PD. This tripartite dissociation of clinical severity accords with hierarchical classification schemes based on clinical features. 17
The factors that determine the routes of α-synuclein propagation remain to be discovered but likely depend in part on individual genetic features. For example, differential patterns of SNCA gene expression may facilitate or impede propagation.
Our studies address criticism #1 above of Surmeier et al. 8 Using ICA or PLS we find that, after the substantia nigra, the most affected brain regions are the other nodes of the basal ganglia (notably striatum), consistent with known connectivity. It remains to be seen why striatal medium spiny neurons appear not to express Lewy pathology, although we suggest that they can transmit the pathology on to the neocortex (see below).
Finally, we tested the theory that propagation to the limbic system and neocortex occurs at later time points. 18 One can make 2 hypotheses: (1) the progression of atrophy to the cortex will be explained by “exposure” to the subcortical “disease reservoir” and (2) this spread to cortex should correlate with the development of cognitive impairment. The affected regions at any stage of the illness represent a disease reservoir and exposure of any other region can be defined as the product of connectivity times atrophy in the target region (Figure 2):
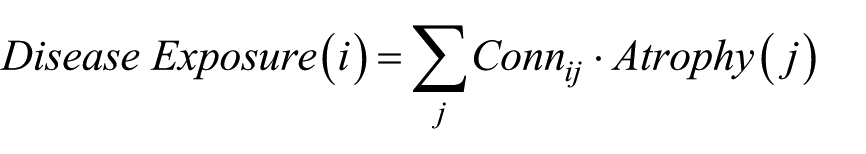
where i refers to the ith cortical region and j refers to the jth subcortical voxel. Atrophy(j) is defined as the z score of atrophy obtained from the DBM analysis from our initial evaluation. 12 Connij is the connectivity between cortical region j and subcortical region i, derived from either functional or anatomical connectomics.
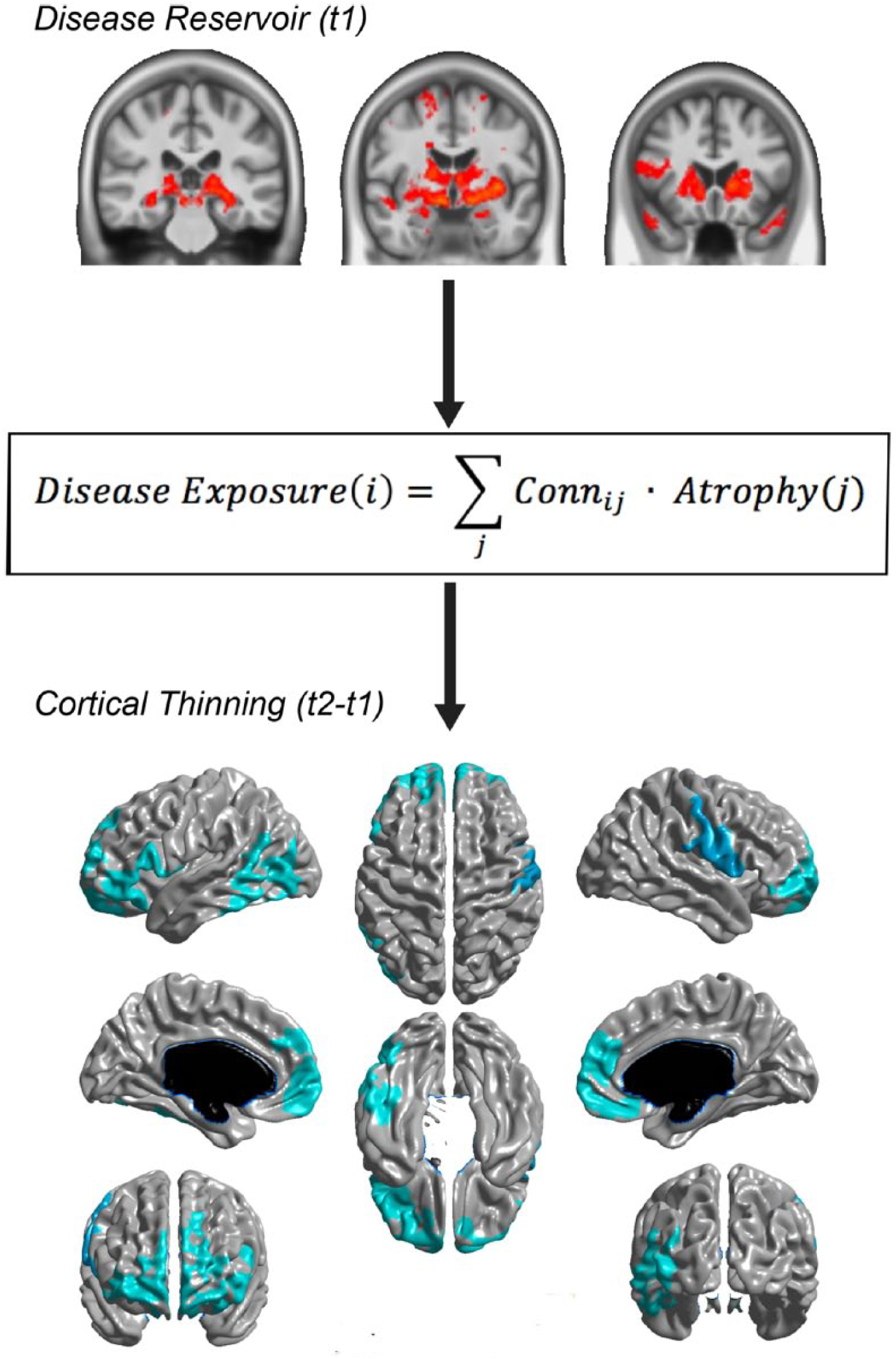
Disease propagation to cortex. Top panel represents atrophy distribution at the first time point (t1), which forms a disease reservoir for subsequent propagation. Exposure predicts cortical thinning at 1 year (t2). Taken from Yau et al. 18
We used follow-up data at 1 year from the PPMI cohort. We found that patients with PD demonstrated greater cortical thinning than controls after 1 year (loss of cortical thickness of 0.3 mm for patients versus 0.2 mm for controls), notably in the medial and lateral ventral frontal cortex, regions belonging to frontoparietal and default mode networks. These ventral frontal areas have also been implicated in certain subtypes of Alzheimer disease 19 and are strongly connected to the basal ganglia. Interestingly, the degree of thinning in these frontal areas correlated with a reduction in cognitive function.
We also found that cortical thinning at each vertex of the cortex was correlated with its disease exposure as defined above, in support of the propagation model. Moreover, this correlation was present whether we defined connectivity based on diffusion-weighted imaging or resting state connectivity. Finally, we found that the best source of disease transmission to the cortex was the basal ganglia, suggesting that, while this region shows little Lewy pathology, it may nonetheless act as a way station for propagation of misfolded α-synuclein to the cortex.
In summary, human neuroimaging data to date support the theory that PD, such as other neurodegenerative diseases, targets intrinsic brain networks. This does not prove the prion hypothesis—other explanations exist. For example, intrinsic brain networks tend to have correlated patterns of gene expression. 20 It is plausible then that selective vulnerability to a global process could lead to atrophy distributions that resemble intrinsic brain networks. Also, brain atrophy could be explained by deafferentation, which would also follow intrinsic connectivity. Nonetheless, coupled with the extensive evidence from cellular biology and postmortem histopathology, the emerging picture is that idiopathic PD is caused by the prion-like propagation of misfolded α-synuclein. Evaluations of the ongoing PPMI data set will be of interest to test the factors that promote or inhibit propagation and how these relate to the natural evolution of symptoms in PD.
Footnotes
Acknowledgements
The author thanks Seyed-Mohammad Fereshtehnejad, Yvonne Yau, Bratislav Misic, Louis Collins, and all the authors of the cited papers for their contributions to this research.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was funded by the research supported by grants from the Canadian Institutes of Health Research, The Michael J. Fox Foundation for Parkinson’s Research, The W. Garfield Weston Foundation, the Alzheimer’s Association, the Natural Sciences and Engineering Research Council of Canada, and a Healthy Brain for Healthy Lives Discovery Award.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
AD and YZ wrote the text.