
Abstract
Background:
Breast phyllodes tumor (PT) is a biphasic tumor and constitutes about 0.3% to 1% of all breast tumors. The PT is histologically classified as benign, borderline, and malignant subtypes. Unlike epithelial breast cancers, PT is derived from breast fibroepithelial tissues, and the genomic information of PT subtypes is still limited.
Objectives:
The objectives were to gain a deeper understanding of genomic changes in the progression of PTs from benign and borderline to malignant.
Design:
In this study, we used an Affymetrix OncoScan Array to analyze the genome-wide copy number variations (CNVs) and nucleotide point mutations from 3 benign PTs, 3 borderline PTs, and 3 malignant PTs collected from the First Affiliated Hospital of Zhengzhou University.
Methods:
DNA was extracted from formalin-fixed paraffin-embedded (FFPE) specimens using the TIANamp FFPE DNA Kit. The DNA was profiled for genome-wide CNV using the Affymetrix OncoScan Array and analyzed using the Nexus Express Chromosome Analysis Suite.
Results:
Our in silico variation analysis indicated copy number loss in Xp11.22 to q22.1 of all benign PTs (χ2 = 9, P = .0027) and 22q11.23 and Xq23 in all malignant PTs (χ2 = 12, P = .0005). A copy number gain was observed in 1p13.3 of all borderline PTs (χ2 = 9, P = .0027) and 7p11.2 of all malignant PTs (χ2 = 9, P = .0027). We also found consistent loss of heterozygosity (LOH) in 32 loci of benign PTs, 32 loci of borderline PTs, and 23 loci of malignant PTs. Among the 87 LOH, there were 15 overlapping loci across all PT subtypes. We observed missense mutations of NRAS, KRAS, IDH2, TP53, and a frameshift deletion in PTEN of sequenced PT samples, irrespective of their subtype. Interestingly, a point mutation in EGFR/EGFR-AS1 was only observed in malignant PTs.
Conclusions:
Our data suggested that CNV at 7p11.2, 22q11.23, and Xq23 together with a point mutation in EGFR/EGFR-AS1 uniquely presented in malignant PTs may correlate with the progression of PTs.
Introduction
Phyllodes tumors (PTs) of the breast are rare neoplasms, accounting for 0.3% to 1% of all breast tumors. The PTs are a heterogeneous group of biphasic fibroepithelial tumors, 1 classified based on their histological characteristics as benign, borderline, and malignant subtypes according to the 2019 WHO Classification of Tumors of the Breast. 2 The current classification of breast PTs is based on histological characteristics, which is challenging for experienced pathologists, and the conclusion for each case is subjective. Because most benign PTs display overlapping characteristics with cellular fibroadenoma, borderline PTs, and some malignant PTs, the field needs to find potential biomarkers to improve the accuracy for PT diagnosis and to monitor disease progression.
In recent years, there have been new findings from genomic studies of PTs.3 -7 These studies, using either whole exome or targeted next-generation sequencing techniques, provided information on genetic alterations in PTs. The somatic mutation of mediator complex subunit 12 (MED12) was frequently observed in PTs.3,4,8 The MED12 is an X-linked gene, and its protein formed a complex along with MED13, CDK8 kinase, and cyclin C, which modulates mediator-polymerase II interactions and thereby regulates transcription initiation and reinitiation rates. 9 Almost all somatic mutations occurred in exon 2 of MED12. The frequency of mutation was higher in benign PTs compared to malignant PTs,10,11 suggesting that MED12 mutations might not be suitable as a biomarker for the malignancy of PTs. Moreover, malignant PTs have been reported to be associated with other somatic mutations in TP53, RB1, and EGFR. 3 Previous genomic studies provided valuable information for the etiology and evolution of PTs. Nevertheless, because of the scarcity of PT samples and the preservation of the majority of PT samples in paraffin, additional studies with sensitive methods to detect genomic changes for PTs are needed in this field.
It is becoming evident that both somatic mutations and genomic copy number changes have roles in the development of various cancer types. 12 Given the small yields of viable DNA extracted from formalin-fixed paraffin-embedded (FFPE) samples, the OncoScan FFPE Assay Kit (OncoScan assay) has been developed to generate whole genome copy number variance (CNV), loss of heterozygosity (LOH), and putative somatic mutation data from as little as 80 ng input DNA from highly degraded FFPE samples. The assay currently detects 74 clinically actionable somatic mutations found in 9 hot-spot cancer genes (TP53, BRAF, PTEN, IDH1, IDH2, EGFR, NRAS, KRAS, PIK3CA) and provides increased copy number (CN) resolution in approximately 900 cancer genes to complement the genome-wide CNV data. 13 Based on Molecular Inversion Probe technology, 14 the genomic probes have a footprint of only 40 bp, optimizing them for binding highly degraded DNA, which is a feature of FFPE samples. 15 Combined analysis of CN and a panel of somatic mutations in a single assay provides a convenient workflow fit for capturing a large portion of the underlying variation driving tumorigenesis and tumor evolution.
To better understand the genetic landscape of PTs, we used the OncoScan assay to analyze genetic profiles including genome-wide CNV, LOH, and acquired somatic mutations of benign PTs, borderline PTs, and malignant PTs. Our results revealed genetic changes in the PT cells that may be informative for the classification of PT, eg, the unique missense mutation of EGFR/EGFR-AS1 only found in malignant PTs in this study. Potentially, our findings may reveal the biology of tumor progression of this rare tumor in the breast.
Materials and methods
Patient samples
In this study, we used the archived surgically removed specimens of breast PTs curated by the pathology department of the First Affiliated Hospital of Zhengzhou University during 2015 and 2016. To confirm the initial diagnosis, 2 additional senior pathologists re-reviewed and classified these PTs to select samples for benign PTs (n = 3), borderline PTs (n = 3), and malignant PTs (n = 3) in accordance with the 2019 WHO classification standard.
Histology of breast phyllodes tumors
Slides from benign PTs, borderline PTs, and malignant PTs were used for histological examination. These slides were sectioned at 5 μm. They were first deparaffinized in 3 washes of xylene (3 minutes each) and then were rehydrated in graded ethanol solutions (100%, 95%, 70%, 50%, and 30%) (1 minute each). The slides were stained with hematoxylin and eosin (H&E). Tumor section images were captured with a KF-PRO-005-EX (KFBio, Zhejiang, China) automatic pathology detection system.
DNA isolation from formalin-fixed, paraffin-embedded phyllodes tumor samples
The FFPE samples, each measuring 5 microns thick, were cut from the selected blocks. Genomic DNA was extracted using the TIANamp FFPE DNA Kit (Tiangen Biotech (Beijing) Co, Ltd, Beijing, China) according to the manufacture’s guidance. The DNA was quantified by Qubit3 Fluorometer (Thermo Fisher Scientific, Waltham, Massachusetts), and its integrity was checked by agarose gel electrophoresis. The DNA was preserved in a plate that was sealed with Micro-Amp Clear Adhesive Film (Applied Biosystems, Waltham, Massachusetts) and kept frozen at −20°C before the subsequent experiments.
The Affymetrix OncoScan chip scan approach
The OncoScan CNV Plus chip (Affymetrix, Santa Clara, California) scan approach was initiated with the extraction of 80 ng genomic DNA from the FFPE PT samples using the molecular-inverted probe (MIP) technique with OncoScan Somatic Mutation Probe Mix and OncoScan Copy Number Probe Mix for the detection of single-nucleotide polymorphism genotyping, insertions, deletions, large fragment CNV, LOH, and somatic mutation. The OncoScan CNV Plus Assay includes standard coverage across the whole genome and high-density CN coverage across 900 cancer genes. Each cancer gene is represented by 20 to 40 probes depending on the length of the gene. The assay also includes a somatic mutation panel covering 64 mutations in 9 genes (TP53, BRAF, PTEN, IDH1, IDH2, EGFR, NRAS, KRAS, PIK3CA). The experiments were carried out in the following major steps. 14 Briefly, the DNA was subjected to hybridization, ligation, first digestion, amplification, and second digestion reactions in a liquid phase. The MIP comprised 7 sequences: 2 endonuclease restriction sites, 2 sequences complementary to the target gene, 2 universal primer sequences, and 1 specific tag sequence. Digested DNA was labeled with biotin. The specific tag sequence of the labeled DNA was hybridized to a chip. The chip was then cleaned, stained using red-fluorescent and green-fluorescent probes, and scanned by GeneChip Scanner3000. Array fluorescence intensity data (CEL files) generated by Affymetrix GeneChip Command Console Software was processed using OncoScan Console software to produce OSCHP files and a set of QC metrics. Genomic CNV and aberrant cell fractions were calculated by the provided “TuScan” algorithm, which infers PT cell content and ploidy from the distributions of the most common CN and germline minor allele frequencies, based on the principles used in Allele-Specific Copy number Analysis of Tumors (ASCAT). 16 The CNVs were analyzed with comparisons with the Database of Genomic Variants (DGV), Online Mendelian Inheritance in Man (OMIM), and other relevant literature. Data were then stratified by High-Confidence somatic mutation calls (as determined by the OncoScan algorithm). Samples were subsequently filtered by OncoScan QC metrics, specifically the Median of Absolute Pairwise Difference (MAPD) and normal diploid SNP QC (ndSNPQC). Those satisfying MAPD ⩽0.3 and ndSNPQC ⩾35 were considered “in bounds” and high quality. Those with MAPD ⩽0.3 and ndSNPQC ⩾26 but < 35 were considered “in bounds” but borderline quality. For each somatic mutation, all samples from PT subtypes were selected from the “in bounds” stratum.
Statistical analysis
R (R Foundation for Statistical Computing, Vienna, Austria) and the statistical software Prism for Windows (version 8, GraphPad Software, Boston, Massachusetts) were used for statistical analysis. The relationship between the CNV frequencies, LOH, somatic mutations, and the different PT subgroups was analyzed using the chi-square test. A P-value of <.05 was considered significant.
Results
Histology of benign, borderline, and malignant phyllodes tumors
The demographic data of the 9 PT patients are listed in Table 1. After H&E staining, we found that the benign PTs showed expansion of the stroma into leaf-like structures (fronds). There was variability of the stromal cellularity, but, in general, the cellularity was low (Figure 1A). For borderline PTs and malignant PTs (Figure 1B and 1C), we adopted the following criteria: marked stromal nuclear pleomorphism; stromal overgrowth; increased mitoses (⩾5 mitoses/mm2; ⩾10 mitoses per 10 high-power fields of 0.5 mm2); increased stromal cellularity, which was usually diffuse; and an infiltrative border. Malignant PT was diagnosed when all the features were present in tumor samples, while borderline PT only presented some features. The summary of detailed clinicopathological data for these PT patients is listed in Table 2.
General information of PT patients.

Histology of PT. (A): benign PT with leaf-like structure (fronds); (B): borderline PT; the tumor border is focally an infiltrative border; (C): malignant PT with stromal nuclei that show moderate to marked nuclear pleomorphism. Bar = 50 µm.
Characteristics of pathological information of PT samples.

Copy number variations of benign, borderline, and malignant phyllodes tumors
Next, we analyzed the CNVs extracted from the FFPE PT samples. We noticed that the frequency of CNV in PTs was significantly higher when their malignancy increased (Figure 2). In benign PTs, copy number loss is the major form of CNV, and it was mainly observed at Xp11.22 to q22.1 (χ2 = 9, P = .0027) (Figure 2A). We did not find gains of copy number in benign PT samples (Figure 2A). With the increased malignancy subtype of PTs, we found that the frequency of gain of copy number was also augmented (Figure 2B and 2C). A gain in copy number was observed at 1p13.3 (where no disease-causing gene exists, per OMIM) in all borderline samples examined (χ2 = 9, P = .0027) (Figure 2B). The frequency of gain of copy number was higher and was comparable to the copy number loss in malignant PTs (Figure 2C). For all the malignant PTs examined, a gain of copy number was observed at 7p11.2 (χ2 = 9, P = .0027). The 7p11.2 cytoband contained 8 genes: VSTM2A, VSTM2A-OT1, SEC61G, LOC100996654, EGFR, EGFR-AS1, ELDR, and LANCL2. Our results showed that copy number loss at 22q11.23 and Xq23 was observed for all malignant PTs but not in benign PTs or borderline PTs (χ2 = 12, P = .0005) (Figure 2C). The 22q11.23 cytoband contained 3 disease-causing genes: LOC391322, GSTT1-AS1, and GSTT1. The Xq23 cytoband contained 12 genes: CHRDL1, PAK3, CAPN6, DCX, LINC00890, ALG13, TRPC5OS, TRPC5, ZCCHC16, LHFPL1, MIR4329, and AMOT. Together, these genomic results indicated that the increase of CNV frequency, especially a gain in copy number, was a characteristic for malignant PTs examined in this study.
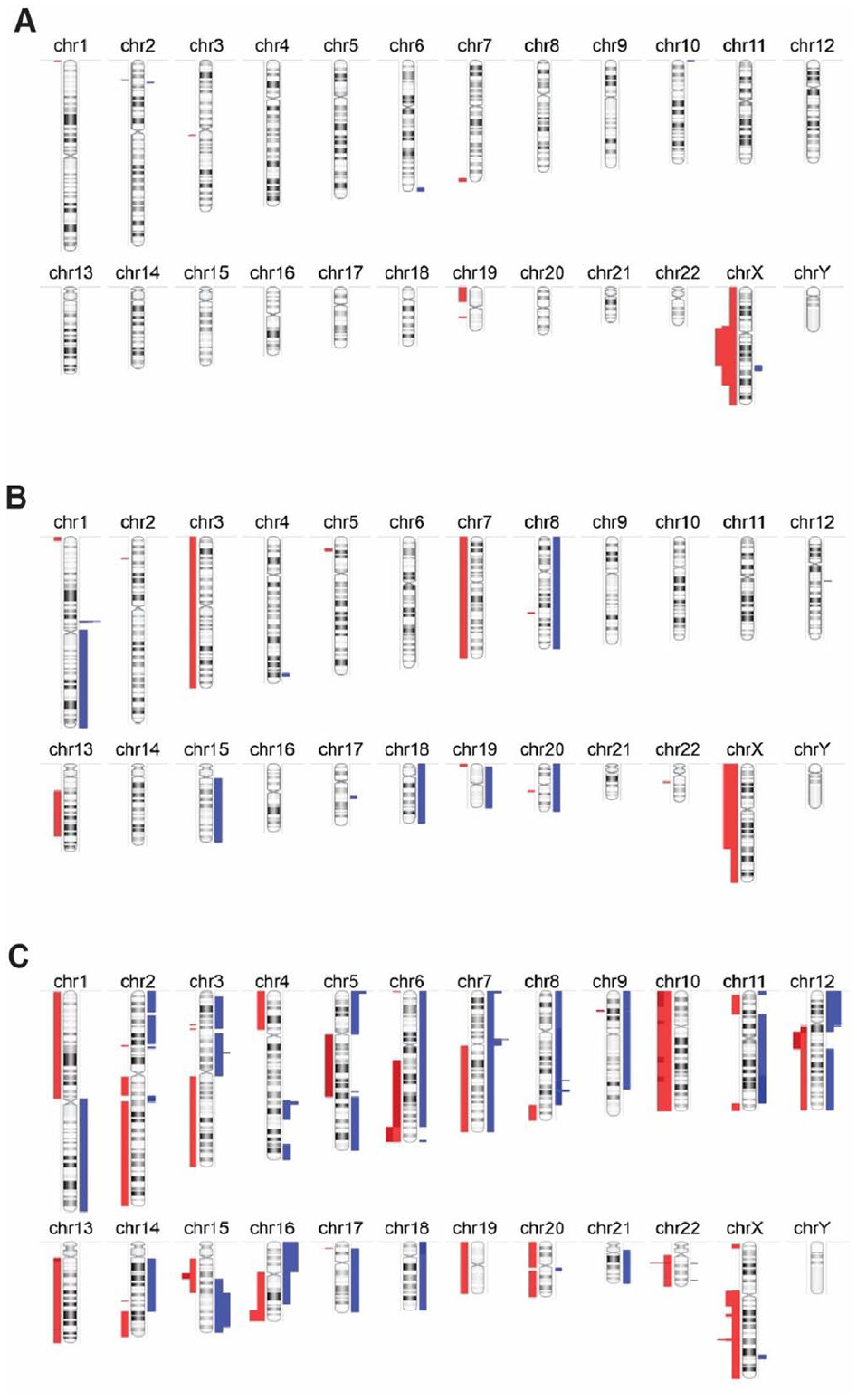
CNV chromosome G-banding of the benign (A), borderline (B), and malignant PTs (C), where blue indicates the gain in CNVs and red indicates the loss of CNVs.
Loss of heterozygosity in benign, borderline, and malignant phyllodes tumors
Next, we analyzed the LOH in PTs. The overall LOH for benign, borderline, and malignant PTs was at similar levels (Figure 3). There were 32 loci in all benign PTs, primarily in chromosomes 1, 2, 3, 4, 5, 6, 7, 9, 10, 11, 12, 13, 16, 17, 19, 22, and X (Figure 3A). Similarly, the 32 loci in all borderline PTs were localized in chromosomes 1, 2, 3, 4, 5, 6, 7, 9, 10, 11, 12, 13, 16, 17, 19, 22, and X (Figure 3B). In malignant PTs, LOH was observed at 23 common loci, primarily in chromosomes 1, 2, 3, 4, 5, 7, 10, 11, 12, 13, 15, 16, 17, 19, 20, 22, and X (Figure 3C). Among these 87 LOH foci, we found 5 LOH loci unique to the benign PTs (primarily in chromosomes 1 and 18), 5 LOH loci unique to the borderline PTs (primarily in chromosomes 2, 5, 6, 11, and 17), and 4 LOH loci unique to the malignant group (primarily in chromosome 5, 15, 16, and 17). Next, we analyzed the common LOH loci between PT subtypes. We found 15 LOH (primarily in chromosomes 1, 2, 3, 5, 7, 9, 11, 13, 16, and X) in all types of PTs. The LOH was observed in 10 loci for both the benign and the borderline groups (primarily in chromosomes 2, 3, 5, 7, 9, 11, 16, and 17). Interestingly, there were only 2 common LOH loci at 19q11 to q13.43 and 20q11.21 to q13.33 in the benign and malignant groups. Similarly, common LOH were observed at 10p15.3 to p11.1 and 11q23.1 to q25 in both the borderline and the malignant PT groups. We summarized the shared and unique LOH in different PT subtypes (χ2 = 41.607, P < .0001), and our data suggested that the 4 unique LOH changes in malignant PTs might come from the increased malignancy of these tumors.

LOH chromosome G-banding of benign (A), borderline (B), and malignant PTs (C).
Somatic mutations in benign, borderline, and malignant phyllodes tumors
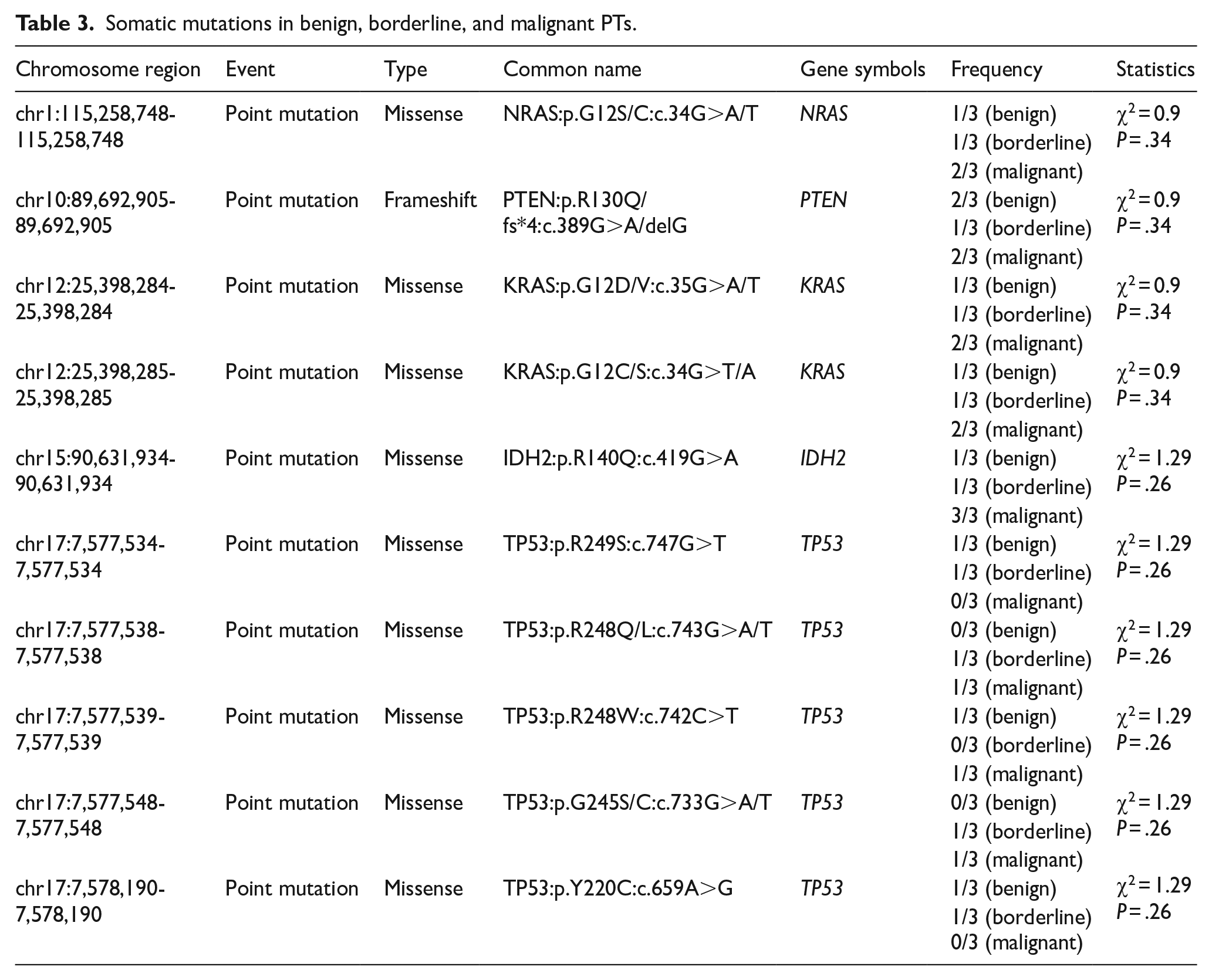
We also noted the missense mutations and sequence variations in all PTs subtypes. We found 9 missense mutations in NRAS, KRAS, IDH2, and TP53, and 1 frameshift deletion mutation in PTEN in benign, borderline, and malignant PTs (Table 3). We found 5 TP53 missense mutations (TP53:p.R249S:c.747G>T; TP53:p.R248Q/L:c.743G>A/T; TP53:p.R248W:c.742C>T; TP53:p.G245S/C:c.733G>A/T; TP53:p.Y220C:c.659A>G) in all PT subtypes, which also comprised most missense mutations found. The TP53 missense R248 hotspot mutation was found in this study and suggested a common mechanism for tumorigenesis of benign, borderline, and malignant PTs. We also identified a unique missense mutation in EGFR/EGFR-AS1 (χ2 = 9, P = .0027, Table 4), which was only observed in all malignant PT samples. Interestingly, the genes of EGFR/EGFR-AS1 localized at 7p11.2, which gained copy number in all the malignant PTs (Figure 2C). These results strongly suggest that the functions of EGFR/EGFR-AS1 might be critical for the transformation of benign/borderline PTs into malignant PTs.
Somatic mutations in benign, borderline, and malignant PTs.
Unique missense mutation in malignant PT.

Discussion
Breast PTs are highly heterogeneous fibroepithelial lesions of the breast. 17 Benign PT is the most common subtype (60%-75%), followed by borderline (15%-26%) and malignant (10%-20%) PT tumors. 2 All 3 PT subtypes have a high probability of recurrence and subsequent grade progression. 18 Of all subtypes, malignant PTs have the highest risk of hematogenous metastatic behavior and may eventually prove fatal. 19 Currently, PT grading based on histological parameters is the most significant prognostic indicator and guide to select the appropriate method for surgical excision. 20 However, the histological grading approach is subjective and needs experienced pathologists to provide conclusions for clinical reference. The accuracy of grade in cases of cross-morphological changes in PTs for pathologists is challenging.2,7 Moreover, the current diagnostic and grading approaches are not ideal to provide accurate prediction of PT development or clear information regarding their recurrence and metastasis. 7 Although several histological studies have attempted to determine prognosis-related tags for PT, 20 translational and clinical studies are needed for more accurate prognostic indicators in PTs.
Breast PTs are a rare cancer compared with other epithelial breast cancers, and the studies of PTs are far fewer. Our study is one of the first to use the archived benign, borderline, and malignant breast PT samples to perform genomic studies with the OncoScan Chip. Our results confirmed that common missense mutations in NRAS, KRAS, IDH2, and TP53 and a frameshift deletion in PTEN were observed in all PTs subtypes (Table 2). More interestingly, point mutations in EGFR/EGFR-AS1 were only seen in malignant PTs (Table 4). These results suggest that mutations in NRAS, KRAS, IDH2, and TP53 contribute to the common tumorigenesis of PTs, while the gain-of-function mutations in EGFR may be closely associated with its malignant progression. A previous report indicated that putative CNVs in TP53, EGFR, and other known cancer driver genes with transforming ability appear only in borderline and malignant PTs but not in benign PTs. 3 This finding from Tan et al is consistent with previous studies showing deregulation of TP53 and RB1 in malignant PTs.21 -23 The discrepancy between our research and others may be that our probe-based sequencing technique is more sensitive to changes with a small amount of DNA than other techniques used previously, 3 and some other researchers21 -23 were only focused on histological analysis of TP53. In summary, our genomic data were highly sensitive and pointed out that mutant TP53 might act as a common driver mutation for tumorigenesis of breast PTs.
Our study reveals some possible markers to predict PT malignancy, including EGFR/EGFR-AS1 and possibly 6 other genes on 7p11.2 with gain of copy number, which may increase their expression in malignant PTs. In the future, we will perform quantitative polymerase chain reaction (qPCR) and histological studies to examine these gene expression levels for predictive markers for PT progression. Previous studies have reported that the gene levels of TP53, Ki67, CD117, EGFR, ET-1, CD10, MMP14, IMP3, and HOXB13 1 ,24 -31 upregulate with increased PT grades. Al-Masri et al 32 and Tariq et al 33 report a positive correlation between CD10 expression and metastasis of malignant PTs. On the contrary, the expression levels of CD34 and ER/PR decrease with increased PT grades. 32 ,34 -36 We found a unique loss of copy number in regions on 22q11.23 and Xq23 in malignant PT samples (see Figure 2). The 22q11.23 cytoband contained no disease-causing genes, while there are 12 genes on Xq23. None of the 12 genes on Xq23 is reported in PTs. Of note, cancer tissues downregulate CHRDL1 expression on Xq23, the loss of which promotes tumor malignancy in breast cancer and colorectal cancer,37,38 and PAK3 expression is positively correlated with a worse prognosis of hepatocellular carcinoma. 39 The loss of function of these genes on Xq23 needs further investigation in future studies of PT progression. Our newly identified potential genes in this study along with previous studies will provide valuable resources to prove their clinical importance for malignant PTs. Other previously identified genetic changes include duplication of chromosome 1q 25 and deletion of 13q, 6p, and 10p, 29 which correlated with tumor progression, tumor grade, and a number of chromosome abnormalities. An array-based comparative genomic hybridization study confirms 9p21 CDKN2A intercalary deletion and 9p deletion in malignant and some borderline PTs, 40 suggesting these genetic changes appear in tumor progression. However, we did not find well-recognized disease-causing genes in the 4 LOH loci that are unique to the malignant group in chromosome 5, 15, 16, and 17 (data not shown). These results suggested that these LOHs might appear after PTs become malignant but not cause the malignant transformation of PTs. In summary, our study along with the previous research will provide a basis to investigate the changes in molecular markers and gene expression profiles associated with malignant PT classification.
Surgery remains the primary treatment modality for malignant PTs. Combinational administration of ifosfamide with doxorubicin has shown effectiveness in some malignant PTs with lung metastasis. 41 Very recently, a genomic study identified an actionable alteration including a TPM4:NTRK1 fusion protein for potential targeted therapy in malignant PTs. 42 Another study reports overexpression of EGFR in more than 90% of malignant PT cases. 43 The EGFR-targeted therapy has been approved by the Food and Drug Administration (FDA) for different types of cancers, especially lung cancer with EGFR mutations. 44 Our finding indicated an additional potential target in EGFR-AS1. This long non-coding RNA mediates EGFR addiction in squamous cell carcinoma. Increasing its expression is sufficient to induce resistance to tyrosine kinase inhibitors. 45 Even though currently there is no targeted therapy in clinical trials for malignant PTs as well as effective prevention of disease progression from benign to malignant PTs, the genomic studies including ours will shed light on the potential for actionable targets in future practice.
We acknowledge that our limitations in having small sample sizes and lacking matched normal tissues might have resulted in the undercalling and overcalling of sequence variants, warranting future studies involving larger cohort sizes with normal tissue samples. We also noticed that because of the limited number of somatic mutation probes provided by the assay kit, we could not detect the mutations in MED12, RARA, and other genes frequently observed by previous studies.3,4,46 Nevertheless, our study provides an independent source for a genomics-based classification of breast fibroepithelial tumors, which may provide useful information to design nucleotide probes, eg, targeting mutant EGFR/EGFR-AS1 to improve the diagnostic accuracy when used in combination with histopathological criteria.
In summary, using a chip scan approach and by searching databases and relevant literature, the present results indicate that copy number gain at 7p11.2 and copy number loss at 22q11.23 and Xq23, and mutation in EGFR/EGFR-AS1 may correlate with malignant progression of PTs. Further analysis will be informative to guide the diagnosis and classification of PT via identification of molecular markers.
Conclusions
In this study, we used an OncoScan FFPE Assay that was designed for small DNA volume from FFPE samples to analyze the genomic changes in benign PTs, borderline PTs, and malignant PTs. Our data indicated unique and shared CNVs, LOHs, and somatic mutations in these tumors. The results also suggested that the mutation in EGFR/EGFR-AS1 might play a role in the progression of benign PTs to malignant PTs. The results from our study provide additional information for the genomic changes in these rare PTs and suggest that EGFR/EGFR-AS1 might be an actionable target to treat malignant PTs.