
Abstract
Chronic diseases impose a global health burden, contributing to high mortality and economic costs. Even with the recent surge in molecular data, these conditions remain largely incurable due to their biological complexity, data fragmentation, and analysis challenges, hindering early diagnosis, mechanistic understanding, and therapy. To address this, we developed PathoAnalyzer-I, an in silico platform that combines bioinformatics and machine learning to decipher chronic diseases. The tool requires no programming skills and provides a user-friendly interface for pathological analysis within a single framework. PathoAnalyzer-I uses a dataset of molecular data for 531 chronic diseases, from databases including the GWAS Catalog, PubChem, and STRING-db, enriched with machine learning–based predictions. Its dual-prediction system enhances molecular insights: one model imputes missing risk alleles with 77.6% accuracy, while a second predicts novel SNP–disease associations with 89.3% accuracy, providing avenues for future research. Applied to Alzheimer’s disease, the platform identified diagnostic biomarkers (e.g. rs6733839T), core genes in disease mechanisms (e.g. BIN1, APOE, SLC24A4, ABCA7, PTK2B), major pathological mechanisms like amyloid processing, synaptic dysfunction, and cellular vulnerability, as well as therapeutic molecules including Beta-Lapachone and preventive compounds such as curcumin. With its features, PathoAnalyzer-I enables scientists, regardless of their resources, to conduct in-depth in silico studies of chronic diseases.
Keywords
Introduction
Despite significant scientific progress in the health sector over recent decades, many chronic diseases remain incurable even with substantial investments aimed at combating them. 1 These conditions impose a considerable burden on multiple levels. They are responsible for millions of deaths worldwide each year and generate staggering economic costs.2,3
The persistence of these diseases is largely attributable to their biological complexity, 4 which hinders an in-depth understanding of their underlying mechanisms. As a consequence, effective prevention and curative strategies remain elusive, and early diagnosis is often challenging.
Alternatively, computational in silico analyses of chronic diseases represent a promising strategy to overcome current limitations. This raises a critical question: despite the availability of numerous high-throughput bioinformatics platforms and multi-omics datasets such as GWAS Catalog, Ensembl, and STRING-DB, why have these resources not been systematically used to characterize the molecular mechanisms that govern chronic pathologies, and to guide their prevention and therapeutic intervention? Several factors may contribute to this gap. First, although molecular datasets including genomics, transcriptomics, proteomics, and metabolomics are increasingly available, they remain highly fragmented and heterogeneous. 5 Currently, no integrative framework offers comprehensive interoperability across these data layers, and the functional links between different molecular entities are rarely captured, which hinders thorough multi-omics integration and analysis. 6 Second, many computational tools are technically demanding, often requiring proficiency in programming, as well as expertise in data normalization, network analysis, and predictive modeling. 7 These barriers limit broader adoption, resulting in the underutilization of resources that could otherwise accelerate mechanistic understanding, biomarker discovery, and the identification of therapeutic and preventive molecules in chronic diseases.
To address these challenges, our team developed PathoAnalyzer-I, a Python-based tool designed to overcome the major obstacles in in silico studies of chronic diseases, including fragmented and disconnected datasets, limited interpretability, and the complexity of existing tools. This all-in-one platform enables scientists, regardless of their resources, to perform comprehensive in silico analyses in a fast and user-friendly manner while generating predictions that open new research directions. By integrating bioinformatics with machine learning, the tool allows researchers to:
Elucidate the molecular mechanisms underlying chronic diseases;
Identify biomarkers for early and confirmed diagnosis
Discover potential therapeutic and preventive molecules;
Explore novel research avenues through machine learning-driven predictions.
Overall, PathoAnalyzer-I enables comprehensive in silico investigations of chronic diseases, facilitating a deeper understanding of their persistence and ultimately contributing to advancements in public health.
Methodology
Theoretical background
Despite the human genome being 99% identical, the crucial 1% of genetic variation holds the key to understanding disease susceptibility. 8 These differences form the core of genome-wide association studies (GWAS), which link specific single-nucleotide polymorphisms (SNPs) to particular diseases and aid in deciphering incurable diseases, including their understanding, treatment, prevention, and early diagnosis. To achieve this goal, several in silico analyses must be undertaken:
- - -
The analysis of these elements necessitates processing tremendous volumes of biological data, which makes automation essential. Bioinformatics supported by tools such as Python, provides efficient solutions through libraries like Biopython 9 and Pandas, 10 enabling accurate and rapid data manipulation.
However, traditional bioinformatics has limitations, especially when addressing chronic diseases that may entail subtle or “silent” biomarkers. In this context, artificial intelligence represents a particularly powerful approach. Machine learning models, such as Random Forest, 11 can generate predictions on SNP-disease associations by leveraging linkage disequilibrium patterns, enabling the identification of potential biomarkers, which help deciphering chronic diseases.
To make this decoding process faster, more accessible, and fully automated, it is essential to bring all these stages together within a unified platform. Such a program would enable users to deeply understand any incurable disease and provide access to potential therapeutic strategies, all through a single, integrated tool. The core principle of our tool is summarized in Figure 1 below.

Core principle of PathoAnalyzer-I.
Dataset
Our methodology was designed to develop a Python program that provides the following functionalities:
Comprehensive disease descriptions for chronic diseases
Analysis of SNPs associated with chronic diseases
Identification and visualization of signaling pathways associated with chronic diseases, with interactive data enrichment
Discovery of potential therapeutic and preventive molecules for chronic diseases
Prediction of novel SNP-disease associations
Raw data
To decode chronic diseases, it is essential to rely on reliable, comprehensive, and accurate data. To meet this need, our program’s dataset was built by integrating high-quality information into a robust system, further enriched with predictions generated by a powerful machine learning model.
First and foremost, this dataset was built based on the GWAS Catalog 12 via web scraping, from which we extracted genomic data associated with 531 chronic pathologies in TSV format. Each file encompasses valuable columns on SNPs, including riskAllele, which contains the SNP ID and risk allele (e.g. in the format rs123456-A); locations, which provides the chromosome harboring the SNP and its precise genomic position (e.g. formatted as 14:45678); pValue, which indicates the statistical significance of the SNPs; mappedGenes, which indicates the gene harboring the SNP; as well as traitName, accessionId, and authors.
Preparing predictions
A central methodological objective of this work is to generate predictive biomarkers of chronic diseases in the form of single-nucleotide polymorphisms (SNPs), by leveraging SNPs already associated with chronic conditions to predict novel candidates under the rationale that genetically proximal variants, as indicated by their genomic positions, may exert similar effects and, through linkage disequilibrium, be linked to the same disorder.
The first step of prediction was the pre-processing of the data, to this end, we followed the procedure below:
Relevant feature selection: RiskAllele, location, mappedGene, and pValue, as these features capture the genomic position of variants and their statistical significance.
Context enrichment: Added the geneticontext (e.g. intron/exon status) using Ensembl 13 via web scraping, serving as a functional and positional indicator for each SNP.
Quality control: Removed approximately 4.07% of SNPs with incomplete annotations.
Field normalization: • Split location into chromosome and position. • Separated RiskAllele into variant (RSID) and riskAllele (A, T, C, G).
During this process, we encountered a limitation: approximately 11.69% of the entries contained only the SNP locus ID without the corresponding risk allele (marked as “?”). This prompted a two-tiered prediction strategy: first, predicting missing risk alleles to complete the dataset; second, using the complete dataset to discover novel SNP–disease associations.
Phase 1. Predicting missing risk alleles
The first phase, aimed at predicting missing risk alleles, is detailed in Table 1, which presents the data sources, engineered features, and model specifications.
Predicting missing risk alleles.

The combination of raw and engineered features provides a biologically grounded framework, linking variants, genes, and pathologies while capturing regional, density, and frequency patterns to predict missing risk alleles. CatBoostEncoder transforms categorical variables into numerical representations while preserving their statistical relationships with the target, and Random Forest leverages these representations to model complex interactions and accurately impute missing risk alleles.
Phase 2. Prediction of novel SNP–pathology associations
The second phase of the framework, which aimed to predict new SNP–pathology associations, is detailed in Table 2.
Predicting new SNP–pathology associations.

To mitigate overfitting, SNPs were divided into groups according to their occurrence frequency (Table 3).
As shown in Table 3, rare variants (⩽4 occurrences) are particularly prone to overfitting due to statistical underrepresentation.
Variant classification by occurrence frequency.

Frequency-based stratification therefore allows to:
Learn group-specific predictive rules
SMOTE to compensate class imbalance
Prevent dominance of common variants in training
Enable stratified cross-validation for unbiased rare-variant evaluation
The two prediction phases were implemented in Python using standard libraries for data manipulation (Pandas, NumPy), 14 machine learning and categorical encoding (scikit-learn, 15 category_encoders 16 ), class imbalance correction imbalanced-learn, 17 and visualization (Seaborn, 18 matplotlib 19 ). Additional utilities were used for file management, logging, and computational support (os, 20 datetime, 21 logging, 22 sys, 23 hashlib, 24 warnings, 25 collections, 26 joblib 27 ).
Model validation
The robustness and generalizability of the machine learning models were assessed using validation frameworks based on simulated genomic data. These validations were conducted without altering the original preprocessing pipelines, feature engineering logic, or hyperparameter configurations, in order to closely reflect real analytical conditions. For the risk allele prediction model, simulated GWAS data comprising 20 000 variants were generated with biologically realistic associations among genomic contexts, genes, pathologies, and nucleotide distributions. Model performance was evaluated using stratified 5-fold cross-validation. A similar validation strategy was applied to the SNP–pathology association prediction model. In this case, 20 000 simulated variants were distributed across five frequency-based categories, ranging from COMMON_VARIANT to ULTRA_RARE_VARIANT, with P-value distributions calibrated to reflect realistic GWAS patterns. Performance was likewise assessed via 5-fold cross-validation. This simulated-data validation phase confirmed the robustness and stability of both models (detailed results are presented in Results: Option 5 – AI-based SNP prediction).
Final dataset
Following these steps, we constructed a clean and robust dataset containing the information required to elucidate the molecular mechanisms underlying chronic and incurable pathologies. Specifically, this dataset comprises 531 files, each named after a chronic incurable pathology and containing two subfolders: the “Reference” subfolder, which includes a TSV file listing GWAS study references associated with the corresponding pathology. Among the key columns in this file are:
The “Pathology” subfolder contains a TSV file with the following columns:
Dynamism of our dataset
Alongside our extensive local dataset, the Python-based program PathoAnalyzer-I incorporates a dynamic data-retrieval system to enhance and update analyses in real time. Data is gathered from reliable and trusted databases, including:
OLS (Ontology Lookup Service): 28 used for detailed pathology descriptions
GWAS Catalog: used for SNP-disease associations
PDB (Protein Data Bank): 29 used for 3D protein structures
Universal Protein Resource (uniprot): 30 used for FASTA sequences and protein functions.
Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) Database: 31 used for gene and protein interaction networks
PubChem Database: 32 used for therapeutic and preventive molecules
Ensembl Genome Browser: used for SNP genomic context
Genotype-Tissue Expression (GTEx) project: 33 used for protein tissue expression data
To ensure fast and efficient performance, we implemented a caching mechanism that streamlines API calls by reducing latency, avoiding redundant queries, and improving productivity.
Another dynamic characteristic of our dataset is its continuous growth, as user queries for previously unrecorded pathologies trigger their automatic inclusion. Indeed, when a new pathology is queried, a dedicated folder is created to store the data retrieved via web scraping. Furthermore, for each newly integrated pathology, key information such as tissue-specific gene expression levels, characteristic SNPs plot, and 3D protein structures is automatically captured during tool operation.
By integrating data from the most trusted databases, machine learning-based predictions, and real-time data enrichment, our dataset brings together all essential molecular data needed to understand the mechanisms behind incurable pathologies, identifies potential therapeutic molecules to fight against them, and facilitates early diagnosis.
General structure of the program
PathoAnalyzer-I is implemented as a modular Python package. Its architecture is organized into core modules:
This modular design ensures maintainability, scalability, and clear separation of concerns.
Our tool, relies on several Python libraries chosen for their performance and ability to process biological data efficiently. For user interaction and display, we employed prompt_toolkit 34 to build interactive command-line applications, PrettyTable 35 to generate tables in text format, and Colorama 36 to add colors to terminal outputs. For web access and automation, the program uses Requests 37 to perform HTTP queries, Selenium 38 for browser automation, and webdriver_manager 39 to manage browser drivers. Regarding data and image processing, Pillow 40 is used for handling pictures, while FuzzyWuzzy 41 supports fuzzy string matching. For visualization and network analysis, Matplotlib and Plotly 42 generate static and interactive graphics, and NetworkX 43 is dedicated to graph and network manipulation. Finally, for data manipulation and numerical computing, Pandas ensures efficient dataset management, and NumPy provides optimized tools for numerical analysis.
Program workflow
Leveraging its extensive and dynamic local dataset, various Python libraries, and its integration with multiple external biological data sources, PathoAnalyzer-I offers a comprehensive suite of analytical tools for chronic disease research.
After the user selects a pathology, the program searches for a locally available TSV file in the dataset that matches the entered name. If no compatible file is found, the program automatically queries the GWAS API to retrieve the corresponding TSV files. Once the data are available, the program displays an interactive menu composed of five options:
Option 1: pathology description
This option displays a detailed description of the selected pathology. The program queries the OLS API to retrieve pathology description. The information is then formatted and displayed in a readable console layout. This option serves as an introduction to the analysis, allowing the user to verify and understand the pathology before exploring its genetic and molecular aspects.
Option 2: SNP characteristics
This option analyzes SNPs associated with the pathology under three distinct scenarios, depending on the available data:

This score uses the same coefficient for two pieces of information—P-value and sample size (normalized by 1000 to ensure comparable scale)—to capture statistical significance, and a higher coefficient for the number of populations carrying the SNP, as this information enables consideration of the SNPs at an international level.
2.

This score does not take into account the number of populations carrying the SNP, as all SNPs are assigned a coefficient of 1.
3.
The most accurate and effective scenario for studying a pathology is the first one, as it uses SNPs shared across multiple populations (at least two). In the absence of this condition, which depends on available data, the program offers the other two particulars’ scenarios to users, which are based on adapted scoring. In all three cases, the program allows SNP visualization in both tabular and interactive graphical formats and provides detailed information for any selected SNP, including genomic context, associated genes, 3D protein structure, and tissue expression profiles.
Option 3: cell signaling pathways
This option extracts genes associated with characteristic SNPs (according to the previously described filter) and displays them in a gene–protein table. The analysis can be further enriched (based on the user’s choice) by generating a protein–protein interaction network using the STRING database, which can be interactively visualized. Users can also explore detailed information for any selected gene.
Option 4: therapeutic molecules
This option focuses on identifying potential therapeutic molecules by retrieving chemical regulators of a selected gene or protein from PubChem. Each candidate molecule is evaluated through a double filter:
After this double filtering, an interactive HTML report is generated to present the accepted molecules (those passing both filters) and the rejected ones (those failing at least one of the two filters).
Option 5: AI-based SNP prediction
This option reads TSV files containing predicted SNPs and their annotations for the selected pathology. The program displays the list of predicted SNPs and allows users to access detailed information for any selected SNP, including genomic context, location, and associated gene.
This analysis follows the workflow illustrated in Figure 2.

PathoAnalyzer-I workflow overview.
Results
By integrating bioinformatics and machine learning techniques, we developed a tool providing a comprehensive framework for in silico analysis of chronic diseases.
The program requires the user to input the name of the target disease, after which it displays an analysis menu, as shown in Figure 3. In cases of incorrect input, the program automatically identifies and suggests the most closely matching valid disease name.

Analysis menu.
Option 1. Pathology description
This option displays a detailed scientific description of the pathology.
For the rest of the features, we will focus on Alzheimer’s disease as a case study:
Option 2. SNPs characteristics
This option provides the characteristic SNPs of the studied pathology (according to the principle explained above).
In the case of Alzheimer’s disease, the top 10 SNPs are: rs6733839T, rs12590654-A, rs11771145-A, rs12151021-A, rs73223431T, rs2830489T, rs2526377-G, rs602602-A, rs871269T, and rs11218343C.
In addition to the tabular display, the user can choose the number of SNPs to display in graphical form (Figure 4).

Characteristic SNPs plot of the genome-wide association study for Alzheimer’s disease.
For a detailed view of each biomarker, users can select an SNP to obtain its information. Taking SNP rs6733839 as an example, PathoAnalyzer-I displays risk allele (T), chromosomal location (chromosome 2, position 127135234), carrier gene (BIN1), variant type (intronic), gene function (the gene encodes a protein which regulates membrane dynamics, endocytosis, amyloid-beta production, and cytoskeletal organization, contributing to neuronal and cellular function), encoded protein information including 3D structure and amino acid sequence, and tissue-specific expression levels illustrated in Figure 5. In addition, the program provides the predicted impact of this mutation, including potential effects on splicing and regulatory elements within the intron.

Tissue-specific expression level of BIN1, the carrier gene for SNP rs41289512.
Option 3. Cell signaling pathways
This option enables the exploration of a gene interaction network associated with the selected pathology.
Figure 6 illustrates a representative gene interaction network underlying Alzheimer’s disease.

Expanded interaction network at threshold 17.
In addition, users can access information on any selected gene: encoded protein, its function, its 3D structure, and amino acid sequence.
Option 4. Therapeutic molecules
PathoAnalyzer-I also identifies potential therapeutic and preventive molecular candidates for chronic diseases. For Alzheimer’s disease, the program proposes several compounds of interest including Dorsomorphin, Beta-Lapachone, Butyrate, and Curcumin.
Option5. AI-based SNP prediction
PathoAnalyzer-I is characterized by the integration of two machine learning models, enabling enhanced decoding of chronic pathologies.
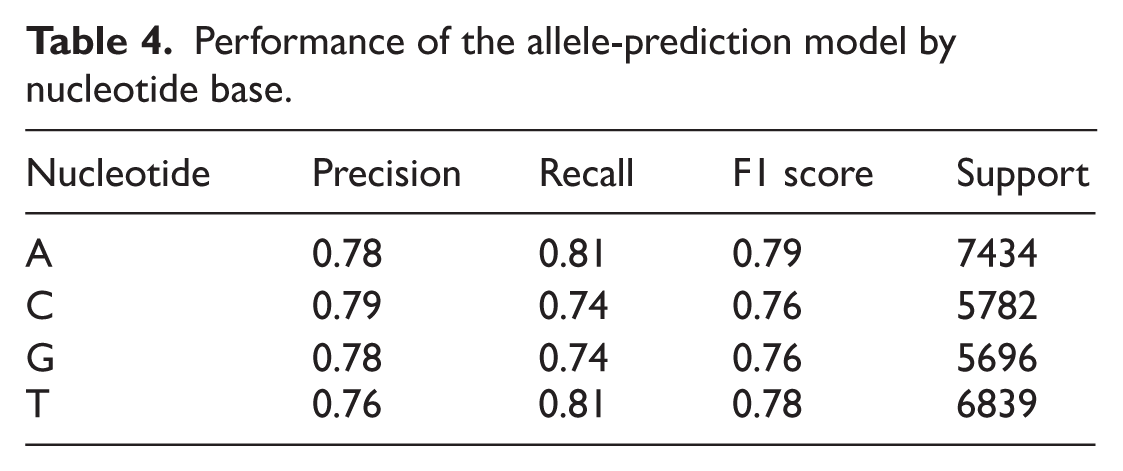
The first model specialized in risk allele prediction achieves 77.6% overall accuracy on real data. Complementary validation on simulated data showed comparable performance with an average accuracy of 75.1% (SD = 0.62%) and a macro-averaged F1 score of 0.730. This agreement suggests that the model maintains stable behavior when applied to new datasets. The detailed performance on real data is presented in Table 4 below:
Performance of the allele-prediction model by nucleotide base.
To illustrate the classification performance and the distribution of prediction errors across nucleotide classes, the corresponding confusion matrix is shown in Figure 7.

Confusion matrix of the risk allele prediction model.
The confusion matrix demonstrates that the model exhibits an overall satisfactory discriminative capability, with the majority of predictions correctly located along the main diagonal, reflecting accurate classification of the four nucleotides. For class A (adenine), 5997 predictions were correct, although notable misclassifications occurred toward T (622 instances) and G (434 instances). Similarly, cytosine (C) was accurately identified in 4267 cases, with errors mainly involving confusion with T (602 cases) and A (511 cases). Guanine (G) achieved 4207 correct predictions while being occasionally misclassified as A (624 cases) and C (354 cases). Finally, thymine (T) yielded 5511 correct classifications, with fewer confusions observed toward A (553 cases) and C (422 cases). Overall, the distribution of errors remained relatively balanced across classes, without evidence of substantial overprediction or underprediction biases. These results are consistent with the precision and recall metrics presented in the performance table (Table 4).
The second model of PathoAnalyzer-I, which specializes in SNP prediction, achieves 89.3% overall accuracy, as detailed in Table 5 below. Validation on simulated data confirmed this robustness with an average accuracy of 87.9% (SD = 0.24%) and a macro-averaged F1 score of 0.831. On simulated data, the model showed excellent discrimination for ultra-rare variants (F1 = 0.985) and common variants (F1 = 0.864). while intermediate frequency classes remained more difficult to distinguish, reflecting the inherent challenge of separating adjacent frequency categories.
Performance of the SNP variant classification model.

To provide a clearer understanding of the classification, the related confusion matrix is presented in Figure 8.

Confusion matrix of the SNP–pathology association prediction model.
The confusion matrix shows that, for the COMMON_VARIANT class, 872 predictions were correct, with 13 instances misclassified as MODERATE_VARIANT. The MODERATE_VARIANT group had 3202 correct predictions, with 38 misclassified as RARE_VARIANT. The RARE_VARIANT category recorded 13 885 correct predictions, with 311 misclassified as VERY_RARE_VARIANT and 105 as ULTRA_RARE_VARIANT. VERY_RARE_VARIANT was correctly predicted in 14 615 instances, with 971 predicted as RARE_VARIANT and 816 as ULTRA_RARE_VARIANT. Finally, the ULTRA_RARE_VARIANT class had 3172 correct predictions, with 1189 misclassified as VERY_RARE_VARIANT.
Taken together, the two validation studies on simulated data showed low cross-validation variability (CV < 3%) and strong out-of-bag agreement (OOB error < 0.1%), indicating minimal overfitting and support the stability of the models. The close convergence between performances obtained on simulated and real data confirms that the models capture genuine biological signals and can be reliably applied to genomic data.
Overall, the PathoAnalyzer-I approach, through the selection of Option 5, enables the identification of novel biomarkers associated with chronic diseases. For Alzheimer’s disease, the tool predicted 2593 SNPs, including rs12916T, rs6859219-A, rs10992900-G, rs192074881-A, rs6498168T, rs1269326T, rs9288520C, rs7671115T, rs7849973T, and rs1044069T. For any predicted SNP, the program provides detailed information; for example, rs192074881-A is located on chromosome 9, within the Fanconi anemia complementation group C (FANCC) gene at position 95 292 358, and is a missense variant.
In addition, the tool predicts missing risk alleles for SNPs already confirmed to be associated with chronic diseases. For instance, the risk allele for rs1060743 (linked to Alzheimer’s disease) 44 was previously unknown (rs1060743-?), but the model successfully predicted it as rs1060743T.
Discussion
Advantages and limitations
Our Python-based software provides an integrated platform for in silico analyses of chronic pathologies.
Designed to optimize computational workflows, it enables rapid data processing and offers an intuitive interface suitable for users with varying levels of expertise.
By combining key analytical modules within a single environment, the tool reduces the need for multiple external applications and supports efficient bioinformatics investigations of chronic disease mechanisms.
Ease of use
Our software is distinguished by its simplicity and ease of use, compatible with all levels of bioinformatics expertise, enabling seamless adoption by researchers. In this regard, Galaxy 45 is another tool known for its user-friendliness, offering an accessible platform for users without programming skills.
Similarly, integrated platforms like the cBio Cancer Genomics Portal (cBioPortal) 46 deliver comprehensive analytical capabilities through intuitive web interfaces. However, leveraging these functionalities typically necessitates navigating multi-layered menus and configuring multistep analytical workflows posing onboarding challenges for users lacking bioinformatics training.
In contrast, command-line frameworks such as Genome Analysis Toolkit (GATK) 47 demand in-depth knowledge of sequencing pipelines and configuration parameters, posing barriers for experimental biologists without computational backgrounds.
Ultimately, ease of use remains a core criterion for selecting in silico analysis tools.
Comprehensive functionality
Integrated bioinformatics platforms have emerged to address the complexity of multi-omics analyses, yet significant gaps persist in chronic disease research. For example, cBioPortal offers robust cancer genomics exploration but provides limited coverage of nononcological chronic conditions and therapeutic insights. In contrast, our software delivers comprehensive analyses of 531 carefully curated chronic pathologies (scalable through program utilization) through a specialized workflow.
DisGeNET 48 operates as a specialized knowledge base for gene-disease associations. While valuable, its framework lacks molecular interaction context essential for mechanistic insights. PathoAnalyzer-I bridges this gap by integrating protein interaction networks and drug discovery within a unified analytical environment.
Where platforms like Open Targets 44 uses multiple modules requiring users to navigate between different contexts, PathoAnalyzer-I offers a streamlined workflow that systematically connects genetic markers to protein networks and therapeutic candidates.
PathoAnalyzer-I’s distinctive architecture integrates six core capabilities:
Pathology Characterization: Comprehensive description of chronic disease.
SNP Characteristics Analysis: Identification and prioritization of pathology-associated SNPs using population-aware scoring strategies, integrating statistical significance and sample size to ensure robust and clinically relevant SNP selection.
Multi-Level SNP Annotation: - Genomic localization and context - Risk Allele - Functional consequences - Associated gene/protein details - 3D protein structure visualization - Tissue-specific expression patterns
Network-Based Mechanistic Analysis: Construction of gene interaction networks using adjustable interaction thresholds.
Therapeutic and Preventive Molecule Screening: Identification of bioactive compounds targeting pathology-associated genes and proteins.
AI-Enhanced SNP Prediction: Prediction of novel SNP–disease associations using machine learning.
In summary, while existing solutions offer valuable specialized capabilities, PathoAnalyzer-I provides an integrative framework specifically optimized for chronic disease research. By combining multi-source data integration with computational predictions in an intuitive interface, we accelerate the translation of genetic insights into actionable therapeutic strategies.
Predictive capability
PathoAnalyzer-I’s two machine learning models demonstrated robust predictive performance and substantial potential to expand both the annotation of SNPs already linked to chronic diseases and the catalog of SNPs associated with these pathologies.
Before discussing the utility of these predictions, it is essential to first address their reliability. Indeed, the missing risk allele prediction model achieved robust discriminative accuracy across all four nucleotides, with an overall accuracy of 77.6%, and demonstrated balanced precision and recall across classes (Table 4). The confusion matrix (Figure 7) further confirms that most predictions were correctly assigned, with adenine (A) and thymine (T) exhibiting particularly high recall (5997 and 5511 correct predictions, respectively), indicating that the model effectively captures nucleotide-specific patterns.
As shown in figure 7, the higher recall for A and T can be partly attributed to the intrinsic nucleotide composition bias of the human genome, which is enriched in A/T bases (~59%) compared to C/G (~41%). 49 This compositional asymmetry biases the model toward A/T bases, explaining the comparatively higher prediction accuracy for these nucleotides. Misclassifications were primarily observed between similar nucleotide pairs (A/T and C/T), reflecting the current limitations of the machine learning model in distinguishing nucleotides with closely related profiles, and suggesting potential avenues for improving feature representation and reducing recurrent errors.
Thanks to the robust accuracy of risk-allele predictions, numerous SNPs that were previously only partially annotated can now be meaningfully exploited. Knowing that an SNP lacking a risk allele is merely a genomic location and cannot be used in risk-allele–dependent analyses, such as those assessing genetic predisposition, this model substantially expands the pool of SNPs available for such analyses by providing accurate risk-allele predictions.
The second machine learning model of PathoAnalyzer-I, specialized in predicting novel SNP–chronic pathology associations, achieved an overall accuracy of 89.3% and demonstrates high precision and recall across all classes (Table 5). The stratification of SNPs by occurrence frequency directly contributed to these results. The confusion matrix (Figure 8) shows that predictions for SNP–pathology associations, relying on SNPs with the closest occurrence frequencies, are more prone to ambiguity due to subtle differences in SNP frequency, which limits the discriminative resolution of the model. Despite these constraints, these results demonstrate the model’s robustness across all classes and ensure that the predictions remain reliable for downstream analyses of chronic diseases.
The predictions generated by the second model can be transformative on several levels.
First, rare chronic pathologies are often characterized by a limited number of validated biomarkers, 50 particularly SNPs, creating substantial ambiguity and delaying progress in their characterization. With PathoAnalyzer-I, however, the set of SNPs associated with these rare diseases becomes sufficiently enriched to support deeper biological insights and accelerate research on their molecular mechanisms.
Second, many chronic diseases are defined by silent biomarkers variants that show no clear involvement in disease pathways. Here, the predictive power of PathoAnalyzer-I is particularly valuable, as it uncovers novel SNP–disease associations that open entirely new avenues for scientific inquiry.
Combined, the predictions from both models, annotation and biomarker identification, provide the scientific community with powerful resources to advance the understanding of chronic diseases and ultimately address their incurability.
Current limitations and challenges of PathoAnalyzer-I
Like most modern bioinformatics platforms, PathoAnalyzer-I leverages distributed data resources to deliver comprehensive analyses. While this architecture ensures exceptional data freshness, it inherently creates dependencies on several external databases, such as the GWAS Catalog, PDB, and UniProt. As a result, when external APIs are temporarily unavailable, certain analytical modules may become inaccessible. To address this, PathoAnalyzer-I incorporates a resilience strategy: Intelligent Caching, whereby frequently accessed datasets (e.g. GTEx expression profiles) are automatically stored locally to reduce external query loads and improve responsiveness. Prospects remain for definitively addressing this limitation in future developments.
Another limitation is that predictive results are not available for pathologies outside the initial dataset, which comprises 531 chronic diseases, as predictions have been precomputed exclusively for this reference set. This limitation is partially mitigated by the fact that this set encompasses a wide range of chronic pathologies. Extending prediction capabilities to additional or newly identified diseases remains a priority for future releases, with plans to integrate real-time prediction modules and further enhance model performance.
The predicted SNPs (Option 5) are probabilistic and statistical in nature and do not represent experimentally validated biological or clinical findings. They should be considered as indicators that depend on the study and population context rather than fixed biological features. In addition, the same allele may be pathogenic in one population and neutral in another. This further confirms the need for in vitro validation to move from statistical inference to biological confirmation.
Furthermore, although PathoAnalyzer-I applies a dual filtering step to candidate therapeutic molecules based on safety and Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) criteria, the safety assessment has certain limitations. While the current list of hazardous GHS codes captures the most critical hazards, further enrichment is required to cover a broader range of safety concerns. In addition, the clinical relevance of these compounds remains to be established. Further in silico validation (e.g. molecular docking and molecular dynamics simulations), followed by in vitro experiments, is required before any biological interpretation or translational application can be considered.
Comparison with existing SNP tools
PathoAnalyzer-I is a Python-based platform for the analysis of chronic diseases, integrating early diagnosis, therapeutic guidance, and signaling pathway investigation. Unlike established online version of tools such as SIFT, 51 PhD-SNP, 52 and I-Mutant 2.0, 53 which focus primarily on the functional or structural consequences of non-synonymous variants, PathoAnalyzer-I implements a disease-centered integrative framework. Specifically, SIFT evaluates amino acid substitutions based on sequence conservation, PhD-SNP predicts the likelihood of an SNP being disease-associated with an accuracy of approximately 74%, and I-Mutant 2.0 estimates mutation-induced protein stability changes, reporting accuracies of 80% and 77% depending on input type. In contrast, PathoAnalyzer-I combines experimentally validated SNP–disease associations from the GWAS Catalog with predictions generated by a machine learning model achieving an accuracy of 89.3%, surpassing the reported performance of PhD-SNP. The platform considers both intragenic and intergenic SNPs, supports gene network analysis, identifies relevant signaling pathways, and proposes potential therapeutic molecules. While a direct comparison with the aforementioned tools is limited by differences in objectives and input data, PathoAnalyzer-I demonstrates superior predictive performance, broader genomic coverage, and a more disease-oriented framework. As a result, it provides a systemic, multi-level view of chronic diseases and enables direct SNP–disease link, offering a more comprehensive and relevant solution for their study.
Deciphering chronic disease
After presenting the structure and functionalities of PathoAnalyzer-I in detail, a central question arises: Does this program truly enable the deciphering of chronic diseases?
In the context of in silico approaches, “deciphering” encompasses several dimensions: pre-diagnosis and diagnosis, understanding the molecular mechanisms underlying a disease, and identifying therapeutic and preventive avenues. To answer this question, we examine the case of Alzheimer’s disease as a concrete example to demonstrate the capacity of PathoAnalyzer-I to decipher chronic pathologies.
Prediagnosis and risk estimation
PathoAnalyzer-I identifies genetic biomarkers (SNPs) that can be exploited to detect genetic predisposition to a disease before the onset of clinical symptoms. This information paves the way for preventive strategies, particularly through the control of gene expression in SNP-carrying genes. Nutritional interventions could achieve this, representing a promising avenue in preventive epigenetics. Unlike simple detection methods, the tool calculates the SNP–pathology association score by taking into account sample size, P-value, and population.
For example, in Alzheimer’s disease, the top 5 SNPs among the 175 meeting the criteria, ranked in descending order, are: rs6733839T, rs12590654-A, rs11771145-A, rs12151021-A, and rs73223431T.
Importantly, probable genetic risk variants converge on key genes, including BIN1, NIFKP9 (rs6733839T), SLC24A4 (rs12590654-A), EPHA1-AS1 (rs11771145-A), ABCA7 (rs12151021-A), and PTK2B (rs73223431T) for which multiple studies have consistently reported significant associations with Alzheimer’s disease.54-57
Finally, several studies highlight the importance of SNPs as early biomarkers of chronic diseases.58-60 In parallel, numerous works emphasize gene expression control as a preventive epigenetic strategy.61,62
Confirmed diagnosis
These biomarkers can also serve in confirmed diagnosis among individuals already presenting clinical symptoms. Based on PathoAnalyzer-I, a subject displaying neurological symptoms in addition to the combination of SNPs (e.g. rs6733839T, rs12590654-A, rs11771145-A, rs12151021-A, and rs73223431T) can be reliably identified as an Alzheimer’s patient. This diagnostic approach is supported by several studies, such as that of Zhang et al. 63
Understanding pathological mechanisms
Cellular signaling is a cornerstone for deciphering pathological mechanisms and identifying metabolites implicated in disease processes. However, in chronic pathologies, well-established signaling pathways and gene interaction networks are often lacking, particularly in Alzheimer’s disease. 64
PathoAnalyzer-I overcomes this limitation by generating a gene interaction network for Alzheimer’s disease (Figure 6). This network highlights 11 central genes BIN1, SLC24A4, ABCA7, PTK2B, CLU, APOE, GRB2, RIN3, NME8, ZCWPW1, CD2AP.
On the other hand, the currently established mechanism of Alzheimer’s disease involves the amyloid precursor protein (APP), which undergoes amyloidogenic cleavage by β- and γ-secretases, producing amyloid-β (Aβ) peptides. These peptides progressively accumulate in the extracellular neuronal space, forming toxic aggregates known as amyloid plaques. Concurrently, tau proteins undergo abnormal phosphorylation, leading to neurofibrillary tangles inside neurons. Together, plaques and tangles trigger early synaptic dysfunction, impairing neuronal communication, and initiate chronic neuroinflammation and oxidative stress. These disturbances ultimately converge on neuronal apoptosis, which underpins the severe cognitive and behavioral symptoms of this chronic disease.65-67
Considering this, the central genes identified by PathoAnalyzer-I play pivotal roles in Alzheimer’s pathogenesis and should be taken into account when interpreting the disease mechanism:
Amyloid metabolism, lipid transport, and immune clearance
APOE, CLU, and ABCA7 regulate lipid homeostasis and amyloid-β (Aβ) dynamics in the brain. The APOE ε4 allele reduces Aβ clearance and promotes aggregation, while CLU acts as an extracellular chaperone controlling Aβ solubility and deposition. ABCA7 contributes to microglial phagocytosis of Aβ and modulates inflammatory responses, thereby linking amyloid accumulation to immune activation.68-70
Endosomal trafficking and APP processing
BIN1, CD2AP, and RIN3 orchestrate endocytic trafficking, APP internalization, and vesicular recycling. Dysfunction of this pathway represents an early pathogenic event in Alzheimer’s disease, favoring amyloidogenic processing and endosomal stress, which precede plaque formation and synaptic impairment.71-73
Synaptic signaling, calcium homeostasis, and tau-related degeneration
PTK2B and GRB2 regulate intracellular signaling pathways essential for synaptic plasticity, neuronal survival, and stress responses, while SLC24A4 maintains calcium homeostasis. In Alzheimer’s disease, GRB2 modulates ERK1/2 signaling and helps protect the neuronal cytoskeleton against β-amyloid–induced damage. Dysregulation of these mechanisms may promote synaptic failure, calcium overload, tau hyperphosphorylation, and cytoskeletal destabilization.74-77
Structural integrity and transcriptional vulnerability
NME8 and ZCWPW1 contribute to cytoskeletal organization and epigenetic regulation of neuronal gene expression. 78 Alterations in these processes may reduce neuronal resilience and amplify vulnerability to neurodegeneration, reinforcing the progressive nature of Alzheimer’s disease.
These mechanisms illustrate that Alzheimer’s pathogenesis emerges from the convergence of amyloid dysregulation, endosomal trafficking failure, immune activation, synaptic dysfunction, and transcriptional vulnerability, each modulated by the key genes APOE, CLU, ABCA7, BIN1, CD2AP, RIN3, PTK2B, GRB2, SLC24A4, NME8, and ZCWPW1. Genetic predisposition, in combination with age-related and environmental risk factors, perturbs the expression and function of these interconnected genes, thereby accelerating pathological onset and promoting progressive neuronal degeneration.
Therapeutic molecules
To date, no curative treatments exist for Alzheimer’s disease. Current strategies aim to alleviate symptoms and slow disease progression.79,80 However, the identification of effective therapeutic strategies is closely linked to a comprehensive understanding of the gene networks and signaling pathways underlying chronic diseases. To achieve this, we must identify regulators for the 11 genes that play a central role in the cellular signaling pathway proposed by PathoAnalyzer-I, as already described—BIN1, SLC24A4, ABCA7, PTK2B, CLU, APOE, GRB2, RIN3, NME8, ZCWPW1, CD2AP—PathoAnalyzer-I proposed gene regulators after a double filtering step (Safety and ADMET), which made it possible to retain potential therapeutic molecules, some of which are summarized in Table 6.
Selected therapeutic modulators of central genes (BIN1, SLC24A4, ABCA7, PTK2B, CLU, APOE, GRB2, RIN3, NME8, ZCWPW1, CD2AP) identified by PathoAnalyzer-I in the context of Alzheimer’s disease.

Some of these molecules, such as curcumin (from turmeric), deguelin (from certain legumes like Derris spp.), and protocatechuic acid (from fruits and vegetables), are of dietary origin, offering preventive potential in addition to therapeutic perspectives.
Another important point concerns Beta-Lapachone, which regulates two key genes (APOE and RIN3) in the identified networks, highlighting a strong potential for therapeutic involvement. Moreover, some studies confirm its therapeutic relevance to Alzheimer’s disease.81,82
The case study of Alzheimer’s disease demonstrates the ability of PathoAnalyzer-I to decipher chronic diseases. The program provides an integrated framework for comprehensive analysis of chronic pathologies, combining molecular-data-driven insights with predictive approaches to support research and translational applications. By uniting multi-source data integration with systematic analyses, the software enables the development of targeted strategies for understanding and managing chronic diseases.
Conclusion
PathoAnalyzer-I is a comprehensive bioinformatics platform designed for the in-depth analysis of chronic pathologies. It integrates large-scale genetic and molecular datasets with advanced machine learning models, enabling precise biomarker annotation, prediction of risk alleles, identification of causal molecular mechanisms, and suggestions of potential therapeutic and preventive interventions. By combining these functionalities within a single environment, the tool provides researchers with rapid, reliable, and holistic insights into disease biology.
The case of Alzheimer’s disease illustrates the capabilities of PathoAnalyzer-I. The platform identified key SNPs, such as rs6733839T, for early risk assessment and confirmed diagnosis, and highlighted central genes including BIN1, APOE, SLC24A4, ABCA7, and PTK2B, which are involved in amyloid processing, synaptic dysfunction, and cellular vulnerability. Moreover, it proposed therapeutic molecules, such as Beta-Lapachone, targeting theses central pathways and dietary compounds, including curcumin, offering preventive benefits through modulation of gene expression. This example demonstrates the platform’s ability to connect genetic data, molecular mechanisms, and intervention strategies in a coherent framework.
Overall, the tool provides the scientific community with a powerful resource to advance the understanding and management of chronic diseases. The platform facilitates comprehensive research into chronic pathologies, ultimately contributing to strategies that improve disease management and public health outcomes.
Supplemental Material
sj-7z-1-bbi-10.1177_11779322261433663 – Supplemental material for PathoAnalyzer-I: An Integrative Bioinformatics Platform for Chronic Disease Analysis
Supplemental material, sj-7z-1-bbi-10.1177_11779322261433663 for PathoAnalyzer-I: An Integrative Bioinformatics Platform for Chronic Disease Analysis by Ali Aguerd, Faiza Bennis and Fatima Chegdani in Bioinformatics and Biology Insights
Footnotes
Ethical Considerations
This study exclusively used publicly available molecular and genomic datasets and did not involve human participants, human tissue, or animals. Consequently, no ethical approval or consent was required.
Author Contributions
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The Python code, datasets, and user guide for the PathoAnalyzer-I platform are publicly available in the Zenodo repository at: ![]()
The materials have been deposited as a 7-Zip archive (PathoAnalyzer-I.7z) containing the following components:
Operating systems: Platform independent (Windows, macOS, Linux)
Programming language: Python
To ensure optimal use of PathoAnalyzer-I, users should:
Install Python and the libraries listed in requirements.txt.
Run the main.py script located in the deciphering_incurable_pathologies folder.
Use English scientific terminology for pathologies, and preferably adopt the nomenclature provided by the GWAS Catalog to ensure consistency and accurate interoperability with both local and external databases.
Maintain a stable Internet connection for reliable and rapid access to external databases.
When using an integrated development environment (IDE) such as PyCharm, a dark theme is recommended, as expressions displayed in light colors may be difficult to read on a light background.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.