
Abstract
Nectin-1/herpes simplex virus glycoprotein D (HSV gD) interaction is crucial to drive herpes simplex virus (HSV) entry. Polyanions are known to show great potential as antivirals. Thus, we explored a peptide-based biotherapeutic approach and, for the first time, evaluated an anionic peptide derived from nectin-1 designed to bind HSV gD. Peptides enriched in acidic and basic residues were selected and computationally modeled using PEP-FOLD3, PROCHECK, ClusPro 2.0, and Desmond. Their antiviral efficacy was tested through virucidal, cell pretreatment, attachment inhibition, entry inhibition, and cytopathic effect (CPE) inhibition assays using a 10 TCID50 (Tissue Culture Infectious Dose 50%) viral dose. Among 4 designed peptides, C1 and C2 showed strong binding to HSV-1 and HSV-2 gD in molecular dynamic (MD) simulations. Peptide C1 exhibited significant virucidal activity (HSV-1: 64.92%, HSV-2: 67.16%), attachment inhibition (HSV-1: 62.03%, HSV-2: 59.38%), and host cell-entry inhibition (HSV-1: 71.37%, HSV-2: 76.28%) at 250 µg/mL concentration. Combination treatment with peptides C1 and C2 at a final concentration of 250 µg/mL (125 µg/mL each) exhibited an additive effect against HSV-1 (68.57%) and HSV-2 (73.37%) infections when tested by CPE inhibition assay. This highlights the potential of HSV gD-targeted anionic peptides for future anti-HSV therapeutics.
Introduction
Herpes simplex virus (HSV) infection is commonly acquired in childhood and remains dormant throughout life. 1 However, in immunocompromised people, the virus may reactivate more frequently, resulting in severe symptoms and consequences such as encephalitis or keratitis. 2 Herpes simplex virus, like other pathogenic viruses, can undergo latent and lytic phases. During the latent phase, neurons and ganglia cells act as the viral reservoir. 3 Commonly prescribed antiviral acyclovir and its analogs are ineffective in controlling HSV infections as the virus can exist in the latent phase.
Compared with the latent phase, the lytic phase of the virus has been extensively studied to develop novel prophylactics or therapeutics. It is widely believed that the most effective approach for developing new antiviral drugs is to target the cell-to-cell spread or initial infection. 4 In the infection phase of the lytic cycle, virus particles attach to the cell’s heparan sulfate proteoglycan (HSPG) receptor through viral glycoproteins gC or gB. Nevertheless, the binding of the virus to HSPG is not permanent or firm. This step facilitates the movement of a virion on the plasma membrane, enabling it to reach neighboring cells or its receptors. 5 Once the virus reaches the vicinity of the receptor (nectin or herpesvirus entry mediator (HVEM)), the virus attempts to enter the cells. 6 From now on, viral cell entry is regulated by gB, gD, gH, and gL glycoproteins. 7 Herpes simplex virus glycoprotein D (HSV gD) binding to its cognate receptor nectin or HVEM or 3-OS-HS causes conformational changes in gD glycoprotein that promote the formation of a fusion complex consisting of gB, gH, and gL. 6 This fusion complex along with the cellular receptors mediates penetration of viral capsid into the host cell through membrane fusion. 8 The gD glycoprotein’s fusion cascade triggering function makes it a promising target for inhibiting HSV’s entry and fusion process. The HSV gD uses nectin-1 as its primary receptor as it is expressed at the site of productive infection. 9 Hence, it is a promising approach to target nectin-1 and HSV gD interaction.
The binding of the virus with its receptor is predicted to be majorly contributed by charge-mediated (ionic) interaction.10,11 The attachment and entry inhibition of various enveloped viruses, such as herpesviruses, retroviruses, orthomyxoviruses, and paramyxoviruses, by polyanionic molecules, highlight the importance of blocking crucial charge-charge interactions. 11 Various synthetic and natural polyanions, including sulphated and sulphonated polysaccharides, carboxylated proteins (such as human serum albumin and milk proteins), sulphated and sulphonated synthetic polyvinylic and acrylic acid polymers, synthetic detergent-amino acid polymers, and polyphosphates (both inorganic and polynucleotide derivatives), have been studied for their potential antiviral activity against human immunodeficiency virus-1 (HIV-1), HIV-2, vesicular stomatitis virus, Semliki Forest virus, Sindbis virus, Influenza A and B, respiratory syncytial virus (RSV), Measles virus, and parainfluenza virus.12-14 These investigations have shown promising results. Polyanionic molecules are predicted to bind with the positively charged basic domains of the virus. They may directly interact with cell-free viral particles 15 or compete with cellular receptors to inhibit viral attachment. 13 Enveloped viruses RSV, HIV, and HSV-2 establish initial contact with the negatively charged HSPGs on the host cell surface.14,16 Therefore, polyanionic molecules are mostly seen to target HSPG. 13 Cyclodextrins and their derivatives have been shown to enhance the antiviral efficacy of various drugs and possess inherent antiviral properties themselves. The β-cyclodextrin (βCD)-based hyper-branched negatively charged polymer, P_PMDA, was observed to disrupt the electrostatic interaction between the host receptor HSPGs and the viral surface protein of HSV-2 and RSV. 17 Similarly, zinc oxide (ZnO) micro-nano structures (MNSs) with partial negative charges, resembling cell-induced filopodia, exhibited anti-HSV-1 activity. Micro-nano structures are expected to selectively bind to cellular heparan sulfate, effectively capturing the virions and inhibiting their entry into human corneal fibroblasts. 18 Moreover, zinc oxide tetrapods (ZnOTs), and zinc oxide tetrapod micro-nanostructures were also reported to neutralize HSV-2 virions with a similar mechanism. 19 Polyanions are also anticipated to hinder virus fusion through direct interaction with the viral fusion peptide (in the influenza virus). Dextran sulfate is a type of sulphated polysaccharide observed to interact with the gp41 fusion peptide of the HIV. 11 Similarly, human serum albumin, a negatively charged protein, has been found to impede the fusion of HIV and influenza viruses.20-22 All these investigations on polyanions emulated that the potential antiviral therapeutic agents should have an anionic (negative) charge.
Protein therapeutics were employed using similar strategies to combat viral infections. The negative charge of an anionic RhoA-derived peptide was found to be significant in demonstrating its antiviral potency against RSV, human parainfluenza viruses, and HIV.10,23,24 In line with this, we have derived peptides from cell surface receptors involved in binding to the virus glycoproteins, so that the virus fails to attach to the cell receptors or cause secondary infections. The basic nature of HSV glycoprotein gD raised the possibility of using anionic peptide as an entry inhibitor. No attempt has yet been made to evaluate an anionic peptide derived from the cytoplasmic domain of the nectin-1 host receptor to target the gD glycoprotein of HSV. Thus, in this study, we have designed and evaluated anionic peptides for their potential to inhibit HSV infection using in silico and in vitro methods.
Materials and Methods
Protein sequence and structures
The nectin-1 sequence was obtained from the UniProt server using the primary accession ID Q15223 (https://www.uniprot.org/). The crystal structures of HSV-1 gD and HSV-2 gD were retrieved from the Protein Data Bank (PDB), with the respective PDB IDs 2C36 and 4MYV. 25
Designing of peptides and evaluation of physicochemical properties
The peptides were designed and screened based on the sequence of the cytoplasmic domain of nectin-1 (Figure 1). The nectin-1 sequence was extracted from the UniProt server available in FASTA sequence format. A series of peptides, ranging from 10 to 20 amino acids in length, were generated and subsequently analyzed for their physicochemical properties (net charge, molecular weight, water solubility, and toxin profile) using the ProtParam, PepCalc, and Toxinpred web servers. 26 Subsequently, the PEP-FOLD3 server was used to generate the 3-dimensional (3D) structure of the sequences. 27 The model with the lowest optimized potential for efficient structure prediction score (sOPEP) energy was chosen for further examination. The peptide structure was refined using the protein preparation wizard feature of the Schrodinger program (Schrodinger LLC).

Schematic presentation of the in silico strategy used for designing anti-HSV peptides.
Peptide and HSV gD protein interaction and its stability study
The peptides were subjected to molecular docking with the HSV gD protein using the ClusPro 2.0 web server (https://cluspro.bu.edu/login.php). Peptide and HSV gD were assigned as ligand and receptor, respectively. 28 The binding site of nectin-1 on the HSV gD was identified to be in patch 1 and patch 2 of HSV gD. The HSV-1 gD consists of 2 surface patches: patch 1 (P23, L25, Q27, F223, T230, V231, and Y234) and patch 2 (R36, V37, Y38, H39, Q132, V214, D215, S216, I217, G218, M219, L220, P221, and R222). Likewise, HSV-2 gD consists of 2 surface patches: patch 1 (P23, L25, D26, Q27, F223, N227, V231, and Y234) and patch 2 (R36, V37, Y38, H39, Q132, R134, P198, V214, D215, S216, I217, G218, M219, L220, P221, and R222) (Figure S1).29,30 The amino acids in these patches were designated as critical residues while docking. The best-docked poses, characterized by more robust interaction patterns and lower binding energies, were determined using the “protein interaction analysis” panel of the BioLuminate (Schrodinger LLC) and HawkDock (Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) calculation), respectively.
The stability and conformational changes of protein-peptide complexes were investigated using the molecular dynamic (MD) simulation (Desmond, Schrodinger) studies. The complex was solvated using the TIP3P water solvent system with an orthorhombic water boundary box. The solvated system was then neutralized by adding sodium or chloride ions and 0.15 M NaCl. The OPLS3e force field was employed to minimize the peptide/HSV gD complex. The NTP ensemble class was employed to screen the minimized complex under conditions of 1 atmospheric pressure and 310 K temperature. The MD simulation was conducted for a duration of 100 ns, and the result was analyzed using a simulation interaction diagram tool. A complete analysis of the simulation results was conducted by interpreting the root mean square deviation (RMSD) plot, protein-ligand contacts in stacked bar charts, ligand-protein interactions in a 2-dimensional (2D) interaction diagram, and radius of gyration (rGyr). The left y-axis and right y-axis illustrate the temporal variation of the RMSD of the HSV gD glycoprotein protein and designed peptide, respectively. A 2D trajectory interaction diagram is used to depict the percentage of simulation time that ligand atoms interact with particular target protein residues. Interactions lasting more than 30% of the simulation duration are classified as weak, while those occurring between 40% and 70% of the simulation time are considered medium. Interactions that persist for more than 70% of the simulation duration are categorized as strong interactions. The stacked bar graph represented the protein-ligand contacts in terms of hydrogen bonds, and hydrophobic, ionic, and water bridge interactions. 31
Peptides
Peptide C1 (10-mer peptide: residues 416-425, LQYPDDSDDE) (Lot: U6326HB280-3/PE2163) and peptide C2 (20-mer peptide: residues 436-456, YEEEEEEEEGGGGGERKVGG) (Lot: U586WHJ250-1/PE2934) with a purity of more than 95% were custom synthesized from GenScript Inc (Piscataway, USA). Peptides C1 and C2, which possess favorable water solubility, were weighed and dissolved in sterile distilled water. Stock peptide solution was diluted in serum-free Dulbecco Modified Eagle Medium (DMEM) (HiMedia, Mumbai, India). Furthermore, a maintenance medium containing 2% fetal bovine serum (FBS) was used to prepare different concentrations of peptides for in vitro assays.
Herpes simplex virus propagation and titration
Herpes simplex virus-1 (Sc-16) and HSV-2 (133953-03) viruses were cultured in Vero cells, derived from African green monkey kidney cells. The cells were maintained in DMEM supplemented with 10% FBS and 1% antibiotic antimycotic solution (Himedia) and incubated at 37 °C in a 5% CO2 condition.
The TCID50 (Tissue Culture Infectious Dose 50%), refers to the dilution of a virus needed to infect 50% of the cell population. The TCID50 calculation is employed to evaluate the infectious dose of the virus. 32 Vero cells were cultured in a 96-well plate (1 × 105 cells/mL density) and virus treatment commenced upon monolayer formation. Serial dilutions of the virus, ranging from 10−1 to 10−10, were prepared in DMEM and incubated with cells for 2 hours. The cytopathic effects (CPE) of each virus dilution on cells were observed for 5 days. The infectious dose of the virus was determined using the Reed and Muench method. 33
The MTT assay
A cytotoxicity study using the MTT assay was conducted to determine the safe concentration of the peptide for antiviral assays. Vero cells were cultured in 96-well plates for 24 hours. The cells were then exposed to peptides at different dilutions (15.62, 31.25, 62.5, 125, and 250 µg/mL) in a maintenance medium. The cells were incubated for a further 48 hours. The culture media was replaced with 100 µL of a 2 mg/mL solution of thiazolyl blue tetrazolium bromide (MTT) (Himedia) and then incubated at 37°C for 3 to 4 hours. To solubilize the formazan crystals, 100 µL of DMSO (dimethyl sulfoxide) was added per well. The untreated cells were used as the control group. The measurement of the well’s absorbance was conducted at a wavelength of 540 nm. A formula (% growth inhibition: 100 − [mean OD of individual peptide-treated group/mean OD of control group × 100]) was employed to compute the percentage of cytotoxicity. The % cell viability was calculated by formula 100 – (% cytotoxicity). 34
Antiviral assays
The antiviral effects of peptides at 10 TCID50 virus challenge dose on various stages of the viral life cycle were determined in the following assays.
Vero cells were seeded in 96-well plates at 1 × 105 cells/mL density and incubated at 37° C for 24 hours. Upon reaching monolayer confluency, cells were washed twice with phosphate buffered saline (PBS) to remove the traces of serum, and the following assays were performed. Peptide concentrations of 15.62, 31.25, 62.5, 125, and 250 µg/mL were used for all the assays in quadruplicate wells, and the entire experiment was repeated thrice with freshly prepared peptide dilutions to get consistent results.
Cytopathic effect inhibition assay
Cells were exposed to a suspension of the HSV-1 or HSV-2 virus prepared in a maintenance medium for 2 hours. Then, the viral suspension was replaced with selected nontoxic concentrations of peptides C1 or C2.
Viral inactivation assay
Herpes simplex virus-1 and HSV-2 were pre-incubated with peptides C1 or C2 for 1 hour at 37°C. Post incubation, the required mixture volume was added to the cells grown in the plates and incubated for 2 hours. Subsequently, cells were rinsed with PBS twice to eliminate any unattached viruses.
Cell pretreatment assay
Cells were exposed to peptide C1 or C2 for 1 hour at 37°C in a humidified CO2 incubator, followed by rinsing with PBS before HSV-1 and HSV-2 infection. Cells were infected with the virus for 2 hours and then cells were washed with PBS twice.
Viral attachment inhibition assay
Plates of the monolayer Vero cells were kept at 4°C for 30 minutes. Then peptide C1 or C2 was added to Vero cells along with HSV-1 or HSV-2 and incubated at 4°C for 2 hours. The cells were then rinsed with PBS 2 times to remove unattached viruses.
Viral entry inhibition assay
The cells were pre-incubated at 4°C for 30 minutes. The 10TCID50 of virus challenge dose of HSV-1 or HSV-2 was added to the culture and incubated for a further 2 hours at 4°C. The cells were washed with PBS twice and incubated with a maintenance medium containing peptides C1 or C2 for 1 hour at 37°C. Cells were washed with PBS.35,36
Later the cells were added with a maintenance medium and incubated at 37°C for 5 days in a CO2 incubator. The experiment was conducted by including positive (Acyclovir (ACV)-treated, 12.5 µg/mL), virus (10 TCID50 virus-suspension-treated), and cell (maintenance media-treated) control groups. Following a 5-day incubation period, the MTT assay was employed to determine the % protection induced by peptide in CPE inhibition, viral inactivation, cell pretreatment, viral attachment inhibition, and viral entry inhibition assays. The % protection was calculated as ([A − B]/[C − B]) × 100, where A represents the peptide-treated infected cells, B represents the virus control, and C represents the cell control.
Statistical analysis
The % cell viability and % protection data were expressed as a mean ± SD and representative of the results of 3 independent experiments. The data were analyzed by using 1-way analysis of variance (ANOVA), followed by the Dunnett posttest. A value of P < .05 was considered to be statistically significant. The half-maximal inhibitory concentration (IC50) and 50% cytotoxic concentration (CC50) values of the peptides were determined by nonlinear regression analysis using Graph Pad Prism, version 8.0.2.
Results
Designing of peptides and evaluation of physicochemical properties
The host receptor protein nectin-1 is composed of 517 amino acids. The extracellular topological domain of nectin-1 consists of amino residues 31 to 355. The transmembrane (TM) domain ranges between 356 and 376, while the cytoplasmic domain spans residues 377 to 517 (https://www.uniprot.org/uniprotkb/Q15223/entry). Owing to the high representation of acidic and basic amino acids, residues 415 to 460 of the cytoplasmic domain of nectin-1 were chosen. Four peptides C1, C2, C3, and C4, that correspond to residues 416 to 425, 436 to 456, 431 to 445, and 421 to 440, respectively were selected based on the sequences with varying net charges and lengths and were subjected to the physicochemical property evaluation.
All the selected peptides exhibited good water solubility. The grand average of the hydropathicity index (GRAVY), was calculated as a negative value, suggesting that the peptides were hydrophilic. The ToxinPred analysis did not predict toxicity in the identified peptides. Figures 2A, 3A, S2a, and S3a display the anticipated tertiary structures of the peptides. The C1 and C2 peptides exhibited a short α-helical fragment and adopted an α-helical conformation on its C-terminal side, while the N-terminal region showed an extended structure. Peptide C3 exhibits a predominantly α-helical conformation throughout its length. Peptide C4 displayed a looping structure characterized by an extended loop conformation between the N-terminal and C-terminal regions. Whereas the AlphaFold-predicted structure of the full-length native nectin-1 reveals that the region encompassing residues 415 to 436 adopts an extended or random coil conformation.

(A) 3D structure and physicochemical properties of the peptide C1 predicted by the PEP FOLD3 server, ProtParam, PepCalc, and ToxinPred. Structures are represented in cartoon input style and color by uniform pattern. (B) Ramachandran plot of the peptide C1 predicted by the PROCHECK server. Red color area (A, B, and L) represent the most favored regions; yellow color area (a, b, l, and p) represent additionally allowed regions; light brown color area (~a, ~b, ~l, and ~p) represent generously allowed regions. (C) The binding modes of peptide C1 to the HSV gD protein. The docking complex of the HSV gD protein with peptide C1 is represented in pose and surface views.

(A) 3D structure and physicochemical properties of the peptide C2 predicted by the PEP FOLD3 server, ProtParam, PepCalc, and ToxinPred. Structures are represented in cartoon input style and color by uniform pattern. (B) Ramachandran plot of the peptide C2 predicted by the PROCHECK server. Red color area (A, B, and L) represent the most favored regions; yellow color area (a, b, l, and p) represent additionally allowed regions; light brown color area (~a, ~b, ~l, and ~p) represent generously allowed regions. (C) The binding modes of peptide C2 to the HSV gD protein. The docking complex of the HSV gD protein with peptide C2 is represented in pose and surface views.
The Ramachandran plot analysis conducted by PROCHECK indicated that the amino acid residues of peptides C1, C2, C3, and C4 were within the acceptable range of allowed regions, and none of the residues being in the disallowed region (Figures 2B, 3B, S2b, and S3b). This result suggests the high quality of the peptide models.
Peptide and HSV gD protein interaction and its stability analysis
The docking investigation was undertaken to identify peptides that interact with the nectin-1-binding sites of HSV gD. The ClusPro docking server produced 4 to 9 poses, with the best pose determined by binding free energy. The binding mode of each peptide with HSV gD protein is shown in Figures 2C, 3C, S2c, and S3c. Peptide C1 binds to Arg222 of the HSV-1 gD protein and Tyr38, His39, Gln132, and Arg134 of the HSV-2 gD protein. Peptide C2 interacted with the critical HSV-1 gD residues Arg222 and Gly218, and HSV-2 gD residues Gln132 and Tyr38. Peptide C3 binds to the critical residues, Ile217 and Arg222 of HSV-1 gD and Arg134 and Arg222 of HSV-2 gD. Peptide C4 binds to the critical residues Thr230 and Pro23 of HSV-1 gD, and Arg134, Gln132, Tyr38, and Val37 of HSV-2 gD.25,29,30 Details of these peptide’s interaction with HSV gD protein are presented in Table 1. These computational findings illustrated that an anionic peptide derived from the cytoplasmic domain of nectin-1 can effectively bind to patches of HSV gD that interact with nectin-1.
Interactions of selected peptides with HSV gD protein were observed during docking studies.
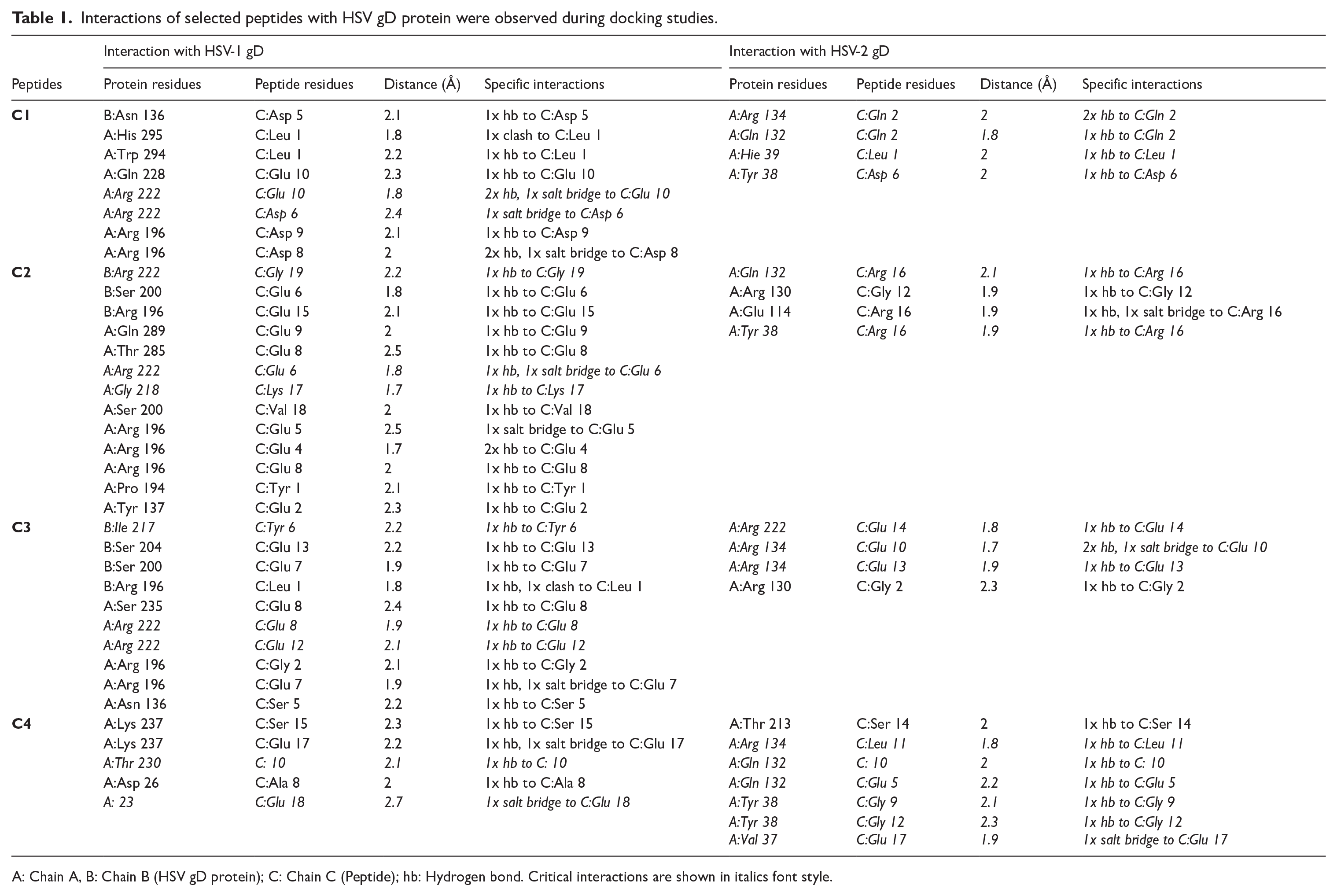
A: Chain A, B: Chain B (HSV gD protein); C: Chain C (Peptide); hb: Hydrogen bond. Critical interactions are shown in italics font style.
The RMSD plot of the peptide C1/HSV-1 gD complex demonstrates stability of the docked complex for a duration of 50 ns. A significant fluctuation occurs between 50 and 60 ns; however, the complex subsequently stabilizes and maintains consistent trajectories for the remainder of the simulation period. Likewise, the peptide C1/HSV-2 gD complex exhibited fluctuations between 20 and 60 ns of the simulation. Nevertheless, it stabilized thereafter and maintained consistent trajectories up to 100 ns within the acceptable range of 1 to 3 Å. The interaction analysis of peptide C1 on HSV-1 gD (stack bar chart) revealed hydrogen bonding, and ionic and water bridge interactions with critical amino residue Arg222 of HSV-1 gD. Also, it predicted hydrogen bonding, hydrophobic interactions, and water bridge contacts with another critical amino residue Ile217 of HSV-1 gD. The C1 peptide formed hydrogen bonding (Val37, Tyr38, His39, Gln132, and Arg134), hydrophobic (Val37 and His39), ionic (Arg134), water bridge contacts (Val37, Tyr38, His39, Gln132, Arg134, Asp215, Met219, and Pro221) with other critical residues of HSV-2 gD (Figure 4A).

RMSD plot and protein-ligand contacts of the (A) Peptide C1. (B) Peptide C2 with HSV-1 and HSV-2 gD protein.
The RMSD plots of the peptide C2/HSV-1 gD and C2/HSV-2 gD complexes display overall stability, with only minor fluctuations remaining within an acceptable deviation range. Peptide C2 formed hydrogen bonding and water bridge contact with critical residues Ile217, Leu218, and Arg222 of HSV-1 gD. Residue Ser216 also exhibited water bridge contact. It formed hydrogen bonding (Val37, Tyr38, Gln132, Arg134, Asp215, and Met219), hydrophobic contacts (Tyr38, His39, Val214, Leu220, and Pro221), ionic interaction (Val37 and Arg134), and water bridge contacts (Val37, Tyr38, Gln132, Arg134, Val214, Asp215, Ser216, Ile217, Gly218, Met219, Pro221, and Arg222) with critical residues of HSV-2 gD (Figure 4B).
Peptide C3/HSV-1 gD complex showed a very stable RMSD plot throughout the simulation. Peptide C3 formed the hydrogen bonding (Arg134, Ser216, Ile217, Gly218, Met219, Leu220, and Arg222), hydrophobic contacts (Ile217 and Leu220), and water bridge contacts (Tyr38, Arg134, Asp215, Ser216, Ile217, Gly218, Met219, Leu220, Pro221, and Arg222) with critical residues of HSV-1 gD. On the other hand, the RMSD plot of peptide C3 in complex with the HSV-2 gD protein exhibited instability, characterized by significant fluctuations throughout the simulation. Peptide C3 formed the hydrogen bonding (Val37, Tyr38, His39, Gln132, and Arg134, Gly218, Met219, and Pro221), hydrophobic contacts (Val37, Leu220 and Pro221), and water bridge contacts (Val37, Tyr38, His39, Gln132, Arg134, Asp215, Gly218, Met219, Pro221, and Arg222) with critical residues of HSV-2 gD. Residues Val37, His39, Arg134, and Asp215 showed ionic interactions (Figure S4a).
The RMSD plot of the peptide C4/HSV-1 gD and peptide C4/HSV-2 gD complexes indicates instability, as it exhibited deviations exceeding 3 Å. Residues Pro23, Leu25, Gln27, Thr230, and Tyr234 of HSV-1 gD exhibited hydrogen bonding and water bridge contacts with peptide C4. It exhibited hydrophobic contacts with Pro23, Leu25, and Tyr234 and ionic interaction with Pro23 residues of HSV-1 gD. Despite unstable RMSD, a very prominent hydrogen bonding and water bridge contact were observed with critical residues of HSV-2 gD. Peptide C4 formed the hydrogen bonding (Val37, Tyr38, His39, Gln132, and Arg134, and Asp215), hydrophobic contacts (Val37, Tyr38, Val214, Pro221), and water bridge contacts (Val37, Tyr38, His39, Gln132, Arg134, Val214, Asp215, Ser216, and Ile217) with critical residues of HSV-2 gD. Residues His39 showed ionic interaction (Figure S4b).
Ligand-protein contacts represented in the 2D interaction diagram revealed that peptide C1 showed strong interaction with HSV-1 gD critical residue Ile217 (72%) and medium strength interaction with Arg222 (43% & 38%). Similarly, peptide C1 demonstrated medium strength interaction with HSV-2 gD critical residue His39 (57%) and strong interaction with Gln132 (92% & 45%). Whereas peptide C2 exhibited weak interactions with HSV-1 gD residue Arg222 (39%) and strong interactions with HSV-2 gD residue Gln132 (73% & 37%). On the other hand, peptides C3 were able to maintain the strong interaction with HSV-1 gD critical residues Arg222 (85% & 87%) and medium strength interaction with Arg134 (61%) residue of HSV-2 gD. Peptide C4 did not show any interactions with critical residues of HSV-1 gD. However, it demonstrated strong interactions with critical residues Tyr38 (78% & 89%), Gln132 (85%), Arg134 (128% & 74%), and Asp215 (88% & 99%) of HSV-2 gD (Table 2).
Description of ligand-protein contacts (2D interaction diagram data) of the peptide/HSV gD complex performed by MD simulation.

Further to assess the structural integrity of the peptide/HSV gD complexes, gGyr was examined over a 100 ns MD simulation. This parameter reflects the overall compactness of the complexes, where higher gGyr values denote a more extended structure, while lower values suggest a tighter, more compact conformation. Among the tested peptides, C1 and C2 complexes with both HSV-1 and HSV-2 gD consistently displayed low and stable gGyr values, indicating strong structural compactness and stability throughout the simulation period. In contrast, peptide C3, though stable with HSV-1 gD, showed significant gGyr fluctuations when complexed with HSV-2 gD. Likewise, peptide C4 exhibited elevated and highly variable gGyr values, pointing toward substantial conformational instability and a less compact complex structure. These findings identify peptides C1 and C2 as the most structurally stable candidates (Figure 5).

Radius of gyration (gGyr) plot of peptide/HSV gD complexes over a 100 ns MD simulation.
Peptide C3 demonstrated favorable interactions with both HSV-1 gD and HSV-2 gD; however, it was not chosen for further analysis because of the unstable RMSD noted in its complex with HSV-2 gD. Peptide C4 was excluded due to its unstable RMSD with both HSV-1 and HSV-2 gD proteins, as well as its inability to interact with the critical residues of HSV-1 gD. Given the objective of identifying peptides capable of effectively targeting both HSV-1 and HSV-2 gD proteins, peptides C1 and C2 emerged as the most promising candidates, supported by their stable RMSD and gGyr profiles and strong binding interactions with critical residues. Thus, only peptides C1 and C2 were chosen for synthesis and subsequent assessment of their antiviral properties.
The MTT assay
A cytotoxicity study was conducted to find out the safe concentrations of C1 and C2 peptides for their use in in vitro antiviral assays. Cell viability of 91.74% and 88.34% at 250 µg/mL peptide concentration, respectively (Figure S5) for C1 and C2 peptides. Both peptides were nontoxic to Vero cells at all the concentrations of 15.62 - 250 µg/mL, and hence these concentrations were selected for these antiviral assays.
Antiviral activities of peptides against HSV-1 and HSV-2 infections
Different cell-based antiviral assays (CPE inhibition assay, viral inactivation assay, cell pretreatment assay, viral attachment, and entry inhibition assay) were used to evaluate the antiviral potential of the peptides C1 and C2. As shown in Figure 6A and B, at the highest concentration (250 µg/mL), peptide C1 demonstrated 49.37% and 48.99% protection against HSV-1 and HSV-2 in the CPE inhibition assay, respectively. In the viral inactivation assay, peptide C1 at 250 µg/mL reduced HSV-1 and HSV-2 infections by 64.92% and 67.16%, respectively, with IC50 values of 109.9 and 111.13 µg/mL (Figure 6C and D). On the other hand, peptide C2 showed minimal antiviral activity in CPE inhibition and viral inactivation assay. Neither C1 nor C2 peptide offered protection to Vero cells in the cell-pretreatment assay (Figure S6a and b).

Antiviral activity of peptides evaluated by (A-B) CPE inhibition assay (C-D) viral inactivation assay at 10 TCID50 viral dose. Virus-infected and ACV-treated cells were used as negative control (Ctr−) and positive control (Ctr+), respectively.
The peptide C1 exhibited dose-dependent activity in CPE inhibition assay, viral inactivation and viral attachment and entry inhibition assays. Peptide C1 offered 62.03% protection against HSV-1 and 59.38% protection against HSV-2 infections at 250 µg/mL in the viral attachment inhibition assay. The IC50 values of peptide C1 against HSV-1 and HSV-2 were 182.93 and 164.6 µg/mL, respectively (Figure 7A and B). Moreover, peptide C1 showed better protection (HSV-1: 71.37%, HSV-2: 76.28%) in viral entry inhibition assay at 250 µg/mL concentration. The IC50 values were found to be 125.86 and 97.76 µg/mL for HSV-1 and HSV-2 infections, respectively (Figure 7C and D).

Antiviral activity of peptides evaluated by (A-B) viral attachment inhibition assay and (C-D) viral entry inhibition assay at 10 TCID50 viral dose. Virus-infected and ACV-treated cells were used as negative control (Ctr−) and positive control (Ctr+), respectively.
Antiviral activity of combinations of C1 and C2 peptides
Equal amounts (125 µg/mL each) of 2 peptides were mixed, and a final concentration of 250 µg/mL of the combined peptide was used for this study. The antiviral experiments, which included cell pretreatment, viral entry inhibition (only against HSV-2), and CPE inhibition assay, showed that the combination of peptide C1 and C2 enhanced the protection compared with the individual active peptide C1. The antiviral activity was significantly increased in the CPE inhibition assay. However, this enhanced activity was observed to be an additive effect of both peptides (Figure 8A and B).

Peptide combination treatment [Peptide C1: 250 µg/mL; Peptide C2: 250 µg/mL; Peptide C1 + C2: final concentration of 250 µg/mL (125 µg/mL each peptide)] against (A) HSV-1. (B) HSV-2 at 10 TCID50 viral dose.
Discussion
In recent decades, the rapid emergence of drug-resistant HSV viruses and the lack of specificity in acyclovir have significantly increased the perceived threat of HSV infections to human life. 37 Peptides are promising candidates for developing novel anti-HSV treatments due to their ease of synthesis and tunability.7,38 The HSV gD protein is a surface glycoprotein essential for its binding to the host cell receptors.30,39 Nectin-1, a cell adhesion protein, consists of 3 extracellular Ig-like domains (V-domain, C1, and C2 domain) and is the primary cognate receptor for HSV gD protein.40-42 The successful entry of the HSV into the host cell depends on the interaction between nectin-1 and HSV gD. 43 The gD protein is a crucial target for peptides that can inhibit the infectivity of HSV, specifically those derived from the nectin-1 host cell receptor protein. As the anionic protein or peptides are known to effectively inhibit viral replication, efforts were made to identify and analyze anionic peptides derived from the cytoplasmic domain of nectin-1 that prevents the entry of HSV into the host cells.
In our in silico docking studies, we observed that the nectin-1-derived peptides C1, C2, C3, and C4 efficiently bind to the nectin-1-binding interface of the HSV gD protein. The amino residues Arg222, Arg134, Gln132, His39, Tyr38, Gly218, Ile217, Thr230, Pro23, and Val37 of HSV gD were identified as being involved in the interaction with the peptide. The MD simulation analysis demonstrated that peptides C1 and C2 substantially kept that crucial contact with both HSV-1 gD and HSV-2 gD. Triazole-pyridines and natural compound (1-(1-benzofuran2-yl)-2-[(5Z)-2H,6H,7H,8H-[1,3] dioxolo[4,5g]isoquinoline-5-ylidene]ethenone) targeting to the HSV-1 gD were reported to bind to these residues of the HSV gD protein.44,45 Our data align with the reported findings and predict the binding of peptides to the nectin-1 binding region of the HSV gD protein. As the best in silico outcome was observed from peptides C1 and C2, the in vitro study was carried out to evaluate the efficacy of this peptide against both HSV-1 and HSV-2 infections.
The antiviral efficacy of peptide C1 was observed in viral inactivation, attachment, and entry inhibition assays against HSV-1 and HSV-2 infections at a virus challenge dose of 10 TCID50. The active peptide C1 demonstrated the highest potency in viral inactivation against HSV-1 (IC50: 109.9 µg/mL) and viral entry inhibition against HSV-2 (IC50: 97.76 µg/mL). The antiviral activity of peptide 1 is likely attributed to its strong interactions with the critical residue Ile217 of HSV-1 gD and HSV-2 residue Gln132. These interactions indicate a more effective binding to the nectin-1 binding interface of the HSV gD viral protein. Whereas peptide C2 exhibited a weaker interaction with the critical residues Arg222 of HSV-1 gD and HSV-2 residues Tyr38, Gln132, Arg134, and Pro221. Moreover, MD simulation results showed that peptide C1 complexes with both HSV-1 and HSV-2 gD consistently exhibited lower and more stable rGyr values compared with peptide C2 complexes. This suggests that C1 forms more structurally compact and stable complexes with the viral protein, which may enhance its inhibitory efficacy. The relative stronger interaction and compact complex formation of peptide C1 compared with peptide C2 suggest adequate binding with the viral protein gD and greater in vitro antiviral activity.
The inability to protect against HSV infection in the cell pretreatment assay and the viral inactivation ability of peptide C1 demonstrate the specificity of the peptide for viral protein rather than the host cell receptor. The addition of peptides during the postinfection stage (CPE inhibition assay) resulted in nearly 50% protection. While the prominent antiviral activity of peptide C1 in inhibiting viral attachment and entry suggests its mode of action. The peptide C1 is anticipated to selectively target the early phases of HSV infection, particularly viral attachment and entry.
To assess whether combinations of peptides targeting HSV gD exhibit synergistic antiviral effects, a mixture of equal amounts of C1 and C2 peptides was evaluated for its antiviral activity. The dual peptide treatment did not exhibit a synergistic effect in any of the antiviral assays, but it did show an additive effect in the CPE inhibition assay. A similar trend was observed with the synthetic peptide P8 (residues 737-756, QYGSFCTAQLNRALSGIAAVEQ) and P9 (residues 890-909, GIGVTAQNVLYENQKQIANQF) targeting the spike (S) protein of the SARS-CoV. Peptide P8 (IC50: 24.9 µg/mL) reduced the SARS-CoV infection and peptide P9 (IC50 > 500 µg/mL) failed to show any significant antiviral activity. 46
The differential activity of peptides C1 and C2 may also be attributed to their respective biophysical properties such as molecular weight and net charge. The peptides C1 and C2 have molecular weights of 1.1 and 2.1 kDa, respectively. Polyanionic molecules with a molecular weight greater than 5 kDa have demonstrated strong inhibitory effects on attachment and entry of various enveloped viruses, such as herpesviruses, retroviruses, orthomyxoviruses, and paramyxoviruses, in vitro.10,11,47 Moreover, the net charge of the polyanion peptide was found to impact its antiviral activity. The RhoA-derived peptide 80 to 94 (a net charge, −3) targeting RSV attachment lost its antiviral activity when its acidic amino acids were replaced by its amide (Peptide 80-94N). The introduction of lysine, a positive charged amino acid residue to RhoA-derived peptides spanning amino acids 77 to 95 results in the loss of their antiviral activity. The data suggest that the negative charge of the RhoA-derived peptide plays a crucial role in determining its effectiveness as an antiviral agent.10,24 In this study, the more effective peptide C1 (with a net charge of −5) is less anionic compared with peptide C2 (with a net charge of −7). The relatively weaker antiviral activity of peptide C2 may be attributed to this factor.
To summarize, peptide C1 demonstrated enhanced antiviral efficacy compared with C2, attributed to its stronger molecular interactions, higher structural compactness and stability, and favorable biophysical characteristics. In addition, detailed investigation of the peptides’ proteolytic stability may further elucidate the reasons behind their differential antiviral effects and presents a valuable direction for future research.
Conclusion
This study focused on the development of a novel anionic antiviral peptide derived from the cytoplasmic domain of the nectin-1 host cell receptor. The in silico findings indicate that peptides C1 and C2 strongly interact with nectin-1-binding patches of the HSV gD glycoprotein. The peptide C1 exhibited the highest potency in inactivating HSV-1 (IC50: 109.9 µg/mL) and inhibiting viral entry of HSV-2 (IC50: 97.76 µg/mL). Peptide C1 which exhibited good antiviral activity can be further modified through a peptidomimetic approach to improve its antiviral therapeutic activity. This study offers valuable insights into the design of anionic antiviral peptides that specifically target the HSV gD protein. The progress in peptide engineering, along with the aforementioned advancements, indicates the potential of these peptides as effective anti-HSV strategies to address significant viral challenges on a wider scale in the future.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322251344130 – Supplemental material for Uncovering the Anti-Herpetic Activity of Anionic Peptides Derived From the Cytoplasmic Domain of Nectin-1
Supplemental material, sj-docx-1-bbi-10.1177_11779322251344130 for Uncovering the Anti-Herpetic Activity of Anionic Peptides Derived From the Cytoplasmic Domain of Nectin-1 by Rakesh Rahangdale, Sumit Birangal, Gautham Shenoy, Fayaz Shaik Mohammad, Mukesh Pasupuleti and Raghu Chandrashekar Hariharapura in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
The authors are thankful to the Indian Council of Medical Research (ICMR), New Delhi, for providing an ICMR-SRF fellowship to RR (File No-5/3/8/56/ITR-F/2022-ITR). Manipal College of Pharmaceutical Sciences, MAHE, Manipal, India, and Manipal-Schrodinger Centre for Molecular Simulation are gratefully acknowledged.
Ethical considerations
Not applicable
Consent to participate
Not applicable
Author contributions
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Intramural fund seed research grant provided by Manipal Academy of Higher Education, Manipal-576104, India [MAHE/DREG/PHD/IMF/2019 dated 7 February 2019, UTN: RG0619259].
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.