
Abstract
Owing to the recent emergence of COVID-19, there is a lack of published research and clinical recommendations for posttraumatic stress disorder (PTSD) risk factors in patients who contracted or received treatment for the virus. This research aims to identify potential molecular targets to inform therapeutic strategies for this patient population. RNA sequence data for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and PTSD (from the National Center for Biotechnology Information [NCBI]) were processed using the GREIN database. Protein-protein interaction (PPI) networks, pathway enrichment analyses, miRNA interactions, gene regulatory network (GRN) studies, and identification of linked drugs, chemicals, and diseases were conducted using STRING, DAVID, Enrichr, Metascape, ShinyGO, and NetworkAnalyst v3.0. Our analysis identified 15 potentially unique hub proteins within significantly enriched pathways, including PSMB9, MX1, HLA-DOB, HLA-DRA, IFIT3, OASL, RSAD2, and so on, filtered from a pool of 201 common differentially expressed genes (DEGs). Gene ontology (GO) terms and metabolic pathway analyses revealed the significance of the extracellular region, extracellular space, extracellular exosome, adaptive immune system, and interleukin (IL)-18 signaling pathways. In addition, we discovered several miRNAs (hsa-mir-124-3p, hsa-mir-146a-5p, hsa-mir-148b-3p, and hsa-mir-21-3p), transcription factors (TF) (WRNIP1, FOXC1, GATA2, CREB1, and RELA), a potentially repurposable drug carfilzomib and chemicals (tetrachlorodibenzodioxin, estradiol, arsenic trioxide, and valproic acid) that could regulate the expression levels of hub proteins at both the transcription and posttranscription stages. Our investigations have identified several potential therapeutic targets that elucidate the probability that victims of COVID-19 experience PTSD. However, they require further exploration through clinical and pharmacological studies to explain their efficacy in preventing PTSD in COVID-19 patients.
Keywords
Introduction
Immediately after being subjected to a strong traumatic event, either experiencing it or witnessing it, a group of people experience a severe stress response (including anxiety, frustration, and insomnia), which could be referred to as posttraumatic stress disorder (PTSD). 1 Although a large number of people go through emotionally disturbing situations throughout their lives, most recover so that they can resume their pretrauma levels of cognitive functioning. 2 Research shows that most people out there are susceptible to traumatic events, although not everyone will develop PTSD. 3 Likewise, approximately 50% to 60% of people may experience traumatic stress during their lives, the lifetime prevalence of PTSD has been calculated at 8.7% only, following Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) codes.4,5 In contrast, several distressing events have been proven to have resulted in PTSD. To exemplify, sexual assaults result in more than 40% of cases of PTSD compared to a lower rate of 5% to 10% witnessed as a result of natural catastrophes.6,7 In general, PTSD rates seem to increase in the presence of domestic violence.8,9 Furthermore, genetic factors influence 30%10,11 to 72% 12 of PTSD threats. Kessler et al 4 have conducted studies and found that genetic factors related to PTSD are also connected to other prevalent psychiatric diseases, namely panic attacks and generalized disorders formed as a result of daily anxieties. Multiple studies have indicated that the serotonin transporter gene (SLC6A4) and a functional variant of FKBP5 increase an individual’s susceptibility to PTSD after a traumatic event.13-15
Four chronic coronaviruses that commonly attack humans in the upper respiratory system have been found to cause symptoms similar to cold or flu. 16 The structural proteins that this virus encodes include membrane proteins, nucleocapsid proteins, envelope proteins, and spike glycoproteins. It also encodes proteins that are not essential parts of it, and most of them contribute to replication and virus transcription processes. 17 In March 2022, the prevalence of syndrome coronavirus 2 (SARS-CoV-2) was increasing globally; noticeably, people in the age group 65 to 74 as well as >75 years experienced the highest increases. 18 Important viral processes must be therapeutically inhibited to reduce the severity of COVID-19. A study found 12 proteins, 8 of which were nonstructural and the rest 4 were accessory, to disrupt cell growth and stability and cause cell death. Among them, only ORF3a was identified as a potential biomarker that could be targeted to fight back against COVID-19. 19
The COVID-19 outbreak is sparked by a national mental health crisis. It is evident that along with the start of COVID-19, there has been an increase in stress response of the general population, use of substances, sadness, anxiety, and suicidal ideation. 20 Descriptive analyses were also conducted, and it was shown that 15.8% of the sample had symptoms of PTSD, 21.6% of the sample had anxiety, and 18.7% had depressed behavior. 21 The meta-analysis carried out by Yuan et al 22 found that the rate of PTSD outbreak was 23.8% among the COVID-19 victims. Individuals who have reported strong connections with a loved one exposed to the virus, reside in COVID-19-affected regions, or have been diagnosed with COVID-19 have an elevated risk of experiencing ambivalent psychosocial functioning; this has been found that increased contact with COVID-19 has been found to have a serious impact on psychology. 21 Prior research has found a number of PTSD calamities that occur from pandemic-induced psychiatric problems.23,24 A study of 602 family members and 307 patients found that kinsmen of COVID-19 victims incorporated a significantly higher prevalence of PTSD symptoms than others. 25
Psychological trauma, caused by the COVID-19 outbreak and the anxiety of infection, can activate the autonomic nervous system and the hypothalamic-pituitary-adrenal (HPA) axis in genetically susceptible individuals. This activation results in the release of glucocorticoids and catecholamines. 26 The immunosuppressive effects of glucocorticoids, which have a range of negative effects on human physiology and behavior, are hypothesized to contribute to PTSD symptoms by altering the HPA axis. 26 Furthermore, patients with PTSD often exhibit weaker immune systems, as evidenced by lower CD8+ T-cell counts. 27 On a separate note, fluoxetine has shown promise in treating depression and contextual fear. Studies by Lu et al 28 demonstrated that long-term treatment with fluoxetine in rats inhibited depression-like behaviors.
The aforementioned findings show that COVID-19 infection and PTSD can share pathological factors and coexist. Previously, a genomic approach was carried out focusing mainly on co-expression and clustering analysis of COVID-19 and psychiatric disorders. 26
This study uses a systems biology approach to identify potential drug target proteins responsible for COVID-19 patients who experience PTSD as an aftereffect. Investigating some original GEO data sets, we identified 15 unique potential drug target proteins and then highlighted their ontological functions, molecular pathways, interactions with miRNA, transcription factors (TFs), repurposable drugs, chemicals, and diseases. To the best of our knowledge, our approach presents cutting-edge approaches capable of defining underlying biological mechanisms in both typical and complex circumstances. To better understand, a briefer idea of the methodologies used in this work is depicted in Figure 1.

Schematic representation of the whole work; starting with collecting the data, taking it through different stages of analysis, and finally trying to provide the clinical specialists with multidirectional data to work with.
Materials and Methods
Collecting the RNA-sequence data sets
The transcriptomic data sets used in this investigation, both for COVID-19 and PTSD, were obtained using the GEO data repository of the National Center for Biotechnology Information (NCBI). 29 Subsequently, the data samples were evaluated using the GREIN portal. 30 We collected a total of 6 data sets with accession numbers GSE64813, GSE109409, GSE114407, GSE150819, GSE164332, and GSE166990 that comprise both case and control samples. Among the data sets, 3 were for PTSD, and the remaining 3 were for SARS-CoV-2. Table 1 provides detailed information considering the number of samples, sources of samples, and other relevant information extracted from the data sets.
Details of information collected from different datasets.

Filtering out less-important data and identifying the DEGs
After collecting the data sets, to filter out the genes that are comparatively less expressed from those that are highly expressed and seem to be potential candidates for both COVID-19 and PTSD, a statistical method of Benjamini-Hochberg was used. 31 As defined by the method, DEGs were identified as those having P values less than .05, and logFC values >1 and <−1. The DEGs for the 6 data sets were identified using the same technique. Later, common differentially expressed genes (DEGs), those shared by the data sets, were identified using the Venny 2.0 Internet server. 32 LogFC and P values of the up-regulated (logFC > 1) as well as down-regulated (logFC < −1) common DEGs were used to draw the heatmaps using the SRplot web server. 33
Using filtered data to form the PPI network and topological matrices to identify the hub proteins
Protein-protein interaction (PPI) is a graphical representation arbitrarily formed comprising nodes (proteins) and edges (proteins’ interactions). 34 Protein-protein interaction network analysis provides the necessary information on proteins and their functions. In our case, the STRING data repository was used to build the network of common DEGs shared by the 2 disease group data sets. 35 Topological adjustments to the network were done by exporting the .tsv file from the STRING database and importing it into Cytoscape. 36 Cytoscape’s cytoHubba plugin’s different topological matrices were used for better identification of the hub proteins. In total, 5 topological matrices were exploited, namely maximum clique centrality (MCC), betweenness, closeness, degree, and edge percolated centrality (EPC). 37 In each matrix, the hub proteins were considered as those with the highest scores, denoting the high protein connectivity between the PPI networks.
Functional and pathway enrichments for the overexpressed gene sets
These analyses are used iteratively to identify enrichments of the overexpressed terms (biological process [BP], cellular component [CC], and molecular function [MF]) and the incorporated pathways by the associated DEGs. A sequence of molecular events having a known start and finish point is referred to as gene ontology (GO):BP. Molecular functions, on the contrary, are defined by the biochemical functions of candidate genes. A gene product is operative in the CC, which is a location in a cell. 38 We conducted our work by investigating the relationships available in the KEGG, Reactome, and WiKi pathways, which are collections of individually produced pathway maps for metabolism, drug development, and other cellular processes. 39 Bubble plots of the common DEGs, which were up-regulated as well as of those that were down-regulated, were drawn using SRplot, which is an online science and research plotting service. Investigations into ontological functions as well as into the enrichments in pathways by the unique hub genes were performed using tools and portals, namely DAVID, 40 EnrichR, 41 Network Analyst, 42 and ShinyGO 43 with GO, 44 KEGG, 45 Reactome, 46 and WikiPathways 47 databases. Chord plots of the gene ontologies and pathways shared by the 4 databases were drawn using the SRplot.
Identification of biomolecules that act as regulators
A regulatory (promotes) and a coding (transcribes) part comprise a gene. Messenger RNAs are formed when the coding part (DNA) goes through the transcription process, and proteins are formed when these RNAs undergo the translation process. The transcription of molecules of an allele is aided by the regulatory part. 48 The expression of genes incorporates a number of complex biochemical processes, including transcription, cooperation, and competition of several TFs, and other mechanisms. Both transcription and expression are significantly altered by transcriptional regulators, namely TFs and miRNAs. 49 Therefore, we exploited the Network Analyst v3.0 platform to identify the regulators’ interactions with unique hub genes, hub genes that were common among the datasets. The web platform also comes in handy to conduct a meta-analysis and transcriptome profiling in addition to gene expression analysis for a variety of organisms. Using the TarBase v8.0 50 and miRTarBase v8.0 51 databases, the gene-miRNA interaction network was evaluated. Then, using the ENCODE 52 and JASPAR 53 databases, the TF-gene interaction networks were constructed. For each database, the degree and betweenness values were adjusted as per requirement.
Evaluation of protein-drug interactions
The underlying properties of ligand affinity are better understood when the correlations of proteins with the available drugs are investigated. 54 Using computational approaches to anticipate particular relationships between them is one option to overcome this knowledge gap. 55 In our case, protein-drug interactions (PDIs) were identified for all interlinked, accountable, and prevalent genes of the 2 diseases studied. Network analyst 42 was used for producing a PDI network by combining the drugs available in the Drug Bank database 56 with the shared unique hub genes.
Investigation of protein-chemistry interactions
The functionality of target biomolecules, largely determined by their interaction partners, is the only way to relate the role of chemical compounds in biological systems. 56 Since diseases sometimes result from many alterations within the same pathway or protein complexes, correlation networks are crucial for the discovery of novel drugs. 57 Protein-chemical interactions are provided by multiple databases, such as proteome-wide interactions or PPI networks,58,59 crucial for in-silico drug discovery. We used the Comparative Toxicogenomics Database (CTD) 60 incorporated within network analyst for investigating chemicals that interact with the unique hub genes.
Analysis of genome-disease associations
The determination of disease genes using in silico methods is crucial for identifying responsible genes and studying genetic disorders. In the past several years, numerous disease-gene prioritization techniques have been created—some basic and several disease-class specific. Very few trustworthy connections are reported to public databases such as GAD 61 and Online Mendelian Inheritance in Man (OMIM). 62 This might be due to the difficulty involved and the lag in human gene-disease investigations. We used the Network Analyst v3.0 online portal along with DisGeNET 63 for the investigations.
Results
Numerical outcomes after DEGs’ analyses and their visualizations
For PTSD, a total of 40 (including 20 cases and 20 controls) samples from peripheral blood leukocytes and peripheral blood mononuclear cell (PBMC) were analyzed. And for SARS-CoV-2, an accumulation of 28 samples (including 13 cases and 15 controls) from bronchi, frontal cortex, and epithelium were analyzed (Table 1). Ignoring duplicates using the Venn diagram, the total number of DEGs for COVID-19 cumulating the 3 data sets, was 2539, and in the case of PTSD, was 945. Furthermore, the DEGs shared between the 2 disease groups was 201 (Figure 2A). Figure 2B and C show 83 up-regulated (logFC > 1) common DEGs, as well as 56 down-regulated (logFC < −1) common DEGs, which were shared between the 2 groups. The heatmap of Figure 2D shows the intensities of the log-fold change and P value ranging from −1 to 1 in a color scale ranging from violet to red for the up-regulated common DEGs, while the heatmap of Figure 2E shows the same for the down-regulated common DEGs.

Venn diagram and heatmap of the DEGs and common DEGs. Venn diagrams: (A) common DEGs between PTSD and COVID-19, (B) DEGs with logFC > 1 and (C) logFC < −1; Heatmaps: (D) DEGs with logFC > 1 and (E) logFC < −1.
Outcomes of investigating the correlation network to filter out the hub proteins
The PPI network was built by first uploading the 201 common DEGs to the STRING database. Then a .tsv file was exported from STRING and imported into Cytoscape to obtain the PPI network. Figure 3 represents the PPI network. In this figure, genes are designated as nodes, whereas links among the nodes are designated as edges. Then we identified Hub genes using Cytoscape algorithms. Mainly the Hub genes were the DEGs ranked in the top 10 among the MCC, betweenness, degree, closeness, and EPC algorithms of Cytoscape’s cytoHubba plug-in (Figure 4). This figure also demonstrates that the hub genes PSMB9, PSMB8, RSAD2, OASL, MX1, as well as 5 others of the HLA group, were shared among the 5 topological matrices. Table 2 shows that HLA-A scored the highest with an MCC score of 1 129 254 having the highest protein connectivity between PPI networks; HLA-B showed the highest scores in terms of the betweenness, degree, and closeness algorithms.

Physical interlinkages between over-expressed proteins. The nodes at the center of each circular-shaped structure represent highly prioritized genes, on the other hand, lines interconnecting them (edges) illustrate the interaction intensities among different genes.

Identification of hub genes using several CytoHubba algorithms. The red-to-yellow hue inclination defines the ranks from high to low.
Topological characteristics of the total unique hub genes identified from several CytoHubba methods.

Outcomes of enrichments’ analyses in functions and pathways by the unique hub genes
The 3 ontological terms’ (namely BP, CC, and MF) analyses were done using the DAVID database, while enrichment analysis of different pathways was performed using the KEGG, Reactome, and WiKi pathways databases incorporated within the DAVID database. The top 10 gene ontologies and pathway terms were tabulated in Tables S1 and S2, respectively. Bubble plots of the gene ontologies and pathways are demonstrated in Figures 5 and 6, respectively. Both figures show that gene ontologies such as the extracellular region, extracellular space, and extracellular exosome, and signaling pathways such as the immune system, adaptive immune system, and the interleukin (IL)-18 signaling pathway incorporate the highest count of DEGs. We identified that 15 genes were uniquely expressed among all 201 common DEGs, shared between the PTSD and COVID-19 datasets. These 15 genes were either common between at least 2 algorithms of MCC, betweenness, degree, closeness, and EPC, or scored among the top 10 in at least 1 of the 5 topological matrices (Table 2). Databases and tools, namely DAVID, Enrichr, Metascape, and ShinyGO were used to perform enrichment analysis of different GO terms and different pathways, and the data collected from the databases were tabulated in Table 3. Chord plots of the gene ontologies and pathways concerning unique hub genes and log fold change values were drawn using the SRplot and are shown in Figures 7 and 8, respectively. The chord plots demonstrate the logFC values in terms of folded chords and in a color scale ranging from violet to red, where violet indicates the lowest change in log fold while red indicates the highest change in log fold value.

Bubble plot representing the enrichments of the significant GO terms.

Bubble plot representing the enrichments of the significant pathways.
Significant GOs and molecular pathways identified from several databases with potential hub proteins.


The chord diagram that illustrates the fold changes of the unique hub genes against the highly enriched GO terms.
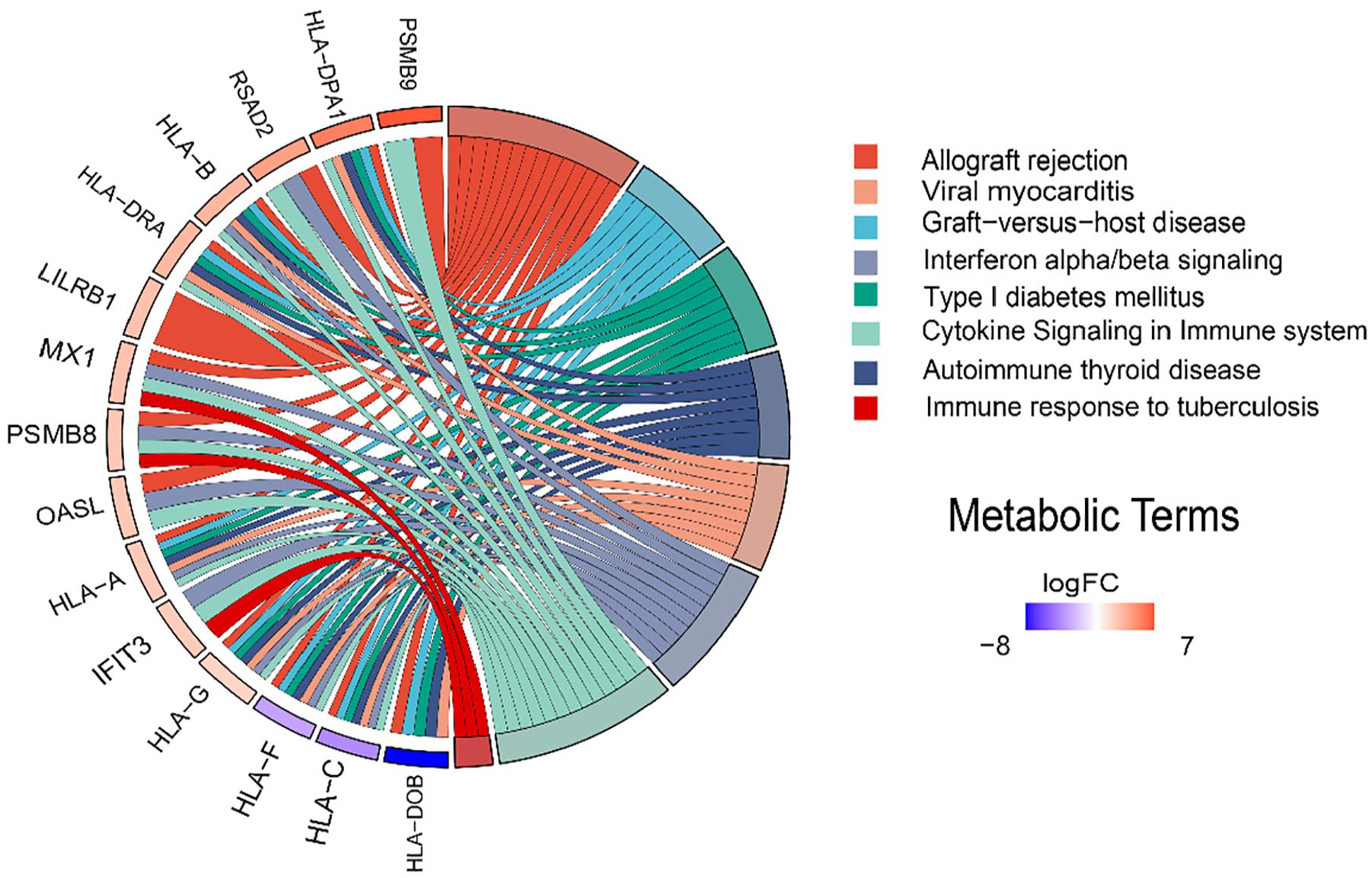
The chord diagram that illustrates the fold changes of the unique hub genes against the highly enriched pathways.
Regulatory network analysis of unique hub genes
Using 2 databases, namely TarBase v8.0 and miRTarBase v8.0, the gene-miRNA interaction network was evaluated. The interaction networks obtained from the 2 databases are visualized in Figure 9A and B, respectively, and the corresponding degree and betweenness scores are tabulated in Table 4. Unique hub genes, namely HLA-A and HLA-C, were found to interact highly with miRNAs such as hsa-mir-124-3p, hsa-mir-146a-5p, hsa-mir-148b-3p, and so on. Furthermore, databases, namely ENCODE and JASPAR, were used to investigate the TF-gene interaction network. The resulting networks are illustrated in Figure 9C and D, respectively. Figure 9C and D together with Table 4 demonstrate that unique hub genes RSAD2, PSMB8, and PSMB9 were densely interacting with TFs such as WRNIP1, IRF1, IRF2, and so on.

Gene-miRNA interaction network using (A) miRTarBase, (B) TarBase databases and TF-gene interaction network using, (C) ENCODE, and (D) JASPAR databases.
Potential transcriptional and posttranscriptional regulators, drugs, chemicals, and disease comorbidities identified at the gene expression level.

Identification of PDIs
To identify repurposable drugs, drugs that are already available in the market, Drug Bank v5.0 incorporated within Network Analyst v3.0 was used. Our investigation, only for the unique hub genes, resulted in a single repurposable drug, namely carfilzomib, which interacts with 2 of the genes, namely PSMB8 and PSMB9 (Figure 10A and Table 4).

(A) Protein-drug interactions using Drug Bank v5.0 database, (B) protein-chemical interactions using the CTD database, and (C) disease-gene association using DisGeNET database.
Investigation of chemical-protein interactions
In-silico drug discovery is largely dependent on the analysis of the chemical-protein interactions (CPIs). In our case, the CTD database, incorporated in Network Analyst v3.0, was used to identify the chemicals that were highly interacting with the unique hub genes. These interactions were illustrated in Figure 10B, and the interaction values based on degree and betweenness score were placed in Table 4. As shown in Figure 10 and Table 4, chemicals such as tetrachlorodibenzodioxin, estradiol, arsenic trioxide (ATO), and valproic acid interacted strongly with unique hub genes, including HLA-G, IFIT3, MX1, and others.
Gene-disease associations’ analysis
The DisGeNET database, incorporated in the Network Analyst v3.0 web server, was used to extract information associating human disease genes and their variants. This information is crucial to identifying disease-responsible genes and analyzing genetic disorders. The DisGeNET-provided interaction network was illustrated in Figure 10C, and the corresponding data were tabulated in Table 4.
Discussion
Integration of network medicine context could help to understand the molecular processes underlying health issues and also for discovering key targets. Transcriptional processes are discovered by RNA sequencing and used to investigate clinical characteristics. 64 We used transcriptome data from COVID-19 patients as well as data from PTSD patients, collected from peripheral blood leukocytes, peripheral blood mononuclear cells, bronchi, frontal cortex, and epithelium to identify possible regulators of COVID-19 that cause PTSD immediately after treatment or at any stages in their lifetime. Now, these data sets are publicly available and are growing in size alongside clinical data sets, so large-scale investigations of interactions between transcript levels and clinical characteristics are possible. However, it was a bit difficult for us to find data sets because limited work has been done on PTSD, particularly at the transcriptomic level. However, we used rich databases like NCBI to find the RNA-seq data sets and analyzed them with another imperial database; named GREIN. However, after analyzing these data sets, we identified shared DEGs and attempted to uncover a variety of potential transcriptomic indicators for disease recurrence. We then employed integrated systems biology approaches to investigate PPI networks, hub proteins, functional and pathway enrichments, gene-miRNA correlations, correlations between proteins, and available drugs, and the link between unique hub genes and other diseases.
In particular, we have explored the vital target proteins and regulatory elements that could be repurposed as drugs for patients who are at a significant risk of experiencing PTSD after being attacked by COVID-19 or treated against COVID-19. Analyzing host gene expression profiles from 3 different datasets for PTSD and 3 for COVID-19, we found that 945 and 2539 genes, respectively, were differentially expressed compared to others. The number of common DEGs between the 2 disease groups was 201. Clearly, these are high or mildly responsible threats in the comorbidity between COVID-19 and PTSD. To investigate further, 5 topological matrices, namely MCC, betweenness, degree, closeness, and EPC of cytoHubba, incorporated in Cytoscape had been exploited to identify the hub genes (top 10% genes of highest connectivity). Hub genes PSMB9, MX1, and HLA-A, -C, -G, and -F were common among the hub genes determined by the 5 different algorithms. A study found that COVID-19 is susceptible to some alleles of the HLA group. 65 The production of some forms of HLA-G is harmful to SARS-CoV-2 infections, was found by another study. 66 A further study found 8 important HLA alleles, commonly carried by PTSD patients. 67
An approach to finding potential biomolecular threats that cause disease is by identifying the biological processes, chemical components, as well as molecular functions mostly influenced by hub genes along with identifying the pathways that incorporate the highest number of hub genes. 68 By merging the hub genes identified by the 5 algorithms, we discovered 15 uniquely expressed genes and used them for further research. Gene ontologies including immune response, cell surface, extracellular exosome, extracellular space, extracellular region, and receptor binding were found to have the highest count of common DEGs. Previously, Cava et al 69 identified ontological terms that are integral and intrinsic to the membrane, and adenyl nucleotide, purine nucleotide, and adenosine triphosphate (ATP) bindings to have the highest count of genes in patients with COVID-19. We found that the pathways, namely the adaptive immune system, immune system, and allograft rejection, incorporated the highest count of common DEGs. Szyda et al 70 found the allograft rejection pathway to be significantly important for resistance to COVID-19 infection. Our findings were strengthened by further analysis of unique hub genes to narrow the direction toward further research, using 4 of the imperial databases. Chord plots of gene ontologies and pathways were used to indicate both up-regulation and down-regulation of logarithmic fold changes of the unique hub genes.
The analysis of gene-miRNA interactions, supported by the data, elucidates that unique hub genes, namely HLA-A and HLA-C, were interacting with miRNAs such as hsa-miR-124-3p, hsa-mir-21-3p, hsa-mir-148b-3p, and so on. A study by Prasad K et al discovered miRNAs, for example, hsa-miR-124-3p as prospective therapy options against COVID-19 along with its related symptoms. 71 Nguyen et al 72 found 74 miRNAs linked to depression, among which hsa-miR-146a-5p was the most significant. Our study investigated genes and miRNAs that were noticeably dysregulated and clarified the putative miRNA-gene regulation network responsible for the comorbidity between COVID-19 and PTSD. Furthermore, TF-gene interaction networks demonstrate that unique hub genes RSAD2, PSMB8, and PSMB9 were densely interacting with TFs such as WRNIP1, IRF1, IRF2, and so on. Lv et al 73 carried out a study that identified TFs, namely WRNIP1, CEBPG, and KLF8, as essential regulators of transcription, in turn, forming the cellular identity of COVID-19. A further study conducted by Breen et al 74 discovered that IRF1, FKPB5, and STAT1 to be potential markers of glucocorticoid stimulation, which is particularly responsible for PTSD.
A single drug, namely carfilzomib, was found to be the repurposable option against the comorbidity of the 2 diseases we studied. Previously, carfilzomib was considered efficient in the treatment of COVID-19-responding patients, especially while caring for elderly people with multiple myeloma. 75 We also noticed that chemicals such as ATO, estradiol, valproic acid, and tetrachlorodibenzodioxin were largely interacting with unique hub genes, namely IFIT3, HLA-G, MX1, and others. Arsenic trioxide can lengthen QTc, increasing the risk of severe arrhythmia and, consequently, COVID-19. 76 Estradiol is renowned for modulating immune cell activities, and therefore, this hormone stimulus can also alter the antiviral defenses of these cells. 77
We found that several diseases including hyperhidrosis disorder, schizophrenia (SCZ), flushing, exanthema, arthralgia, and so on were highly associated with the HLA-A, HLA-B, and PSMB8 genes. This study is important in the area of genetic informatics as candidate gene analysis has been a pioneer in finding risk factors and their correlation with clinical characteristics. 78 Schizophrenia is known to co-occur often with PTSD and is considered the most incapacitating mental illness. 79 Patients recently diagnosed with SCZ and hyperactivity disorder were also shown to have an increased prevalence of COVID-19. 80 Studies have identified further associations between COVID-19, PTSD, and other mental disorders, including major depression disorder (MDD) and childhood mental disorders. Baranova et al and Chen et al, in 3 separate studies, demonstrated that PTSD, MDD, and childhood mental disorders have notable positive genetic correlations with SARS-CoV-2 infection, hospitalized COVID-19, and critical COVID-19.81-83 The study conducted by Chen et al 83 pointed out that attention-deficit hyperactivity disorder (ADHD) again has a noteworthy positive genetic correlation with the 3 COVID-19 traits mentioned above. Finally, researchers have also discovered an increased risk of depression and anxiety in COVID-19 frontline workers. 84 Some comorbid conditions of SCZ, namely type 2 diabetes (T2D), smoking, drinking alcohol, as well as obesity lead to an increased risk of COVID-19.85-87 We found that interferon-stimulated genes, including (ISG) IFIT3, OASL, and MX1, activated by cytokine-like interferons, act as key factors in the development of SCZ and COVID-19. Another study found that excessive release of IL-6 might have a detrimental impact on COVID-19. 88 However, there are cytokines such as CXCL11, CCL4, and interferon (IFN)-γ, that play protective role against COVID-19. 89 Furthermore, it is assumed that genes controlled in different regions greatly influence highly in propagation of complex disorders, for example, PTSD. Despite the important hereditary basis of numerous aberrant neurodevelopmental diseases, COVID-19 may resurface on a consistent frequency, allowing for additional research on diagnosis as well as treatment methods.
The information provided above suggests that our method can reveal basic mechanisms involved in disease pathogenesis, provide new perspectives on cause of the disease, and discover new biomarkers. However, further research is necessary to validate these outcomes. The integration of genetic transcription with functional genomic information can disclose unanticipated medical correlations, potentially providing a novel understanding of neuro and inflectional morphology. Finding the molecular connections between COVID-19 and PTSD could be beneficial for developing promising therapeutic targets. This study may be useful to clinicians, scientists, and many others. It may provide novel opportunities for clinicians to make decisions with the progression of bioinformatics analysis, such as possible hazard assessments, disease identification, and use regular expressions, medication therapy, and dose determination, which is a step toward more to the emergence of completely rejuvenating medicine.
Although the study investigates some authentic GEO data sets of COVID-19 and PTSD cases, the sample sizes incorporated in these data sets range from 6 to 20. To be more specific, their size is somewhat low for definitive conclusions. Investigating with such smaller data sets may also sometimes lead to false-positive outcomes. Therefore, further investigations with larger sample sizes or additional clinical research might be beneficial to validate the significance of our findings.
Conclusion
The work exploited a systems biology approach for identifying drug target proteins responsible for the comorbidity between COVID-19 and PTSD. We identified 15 unique potentially potential drug target proteins and then highlighted their ontological functions, molecular pathways, interactions with miRNA, TFs, repurposable drugs, chemicals, and diseases. However, additional clinical research is advised to confirm the therapeutic targets and correlational and pathobiological factors that underlie the comorbidities between COVID-19 and PTSD disease.
Key Points
Therapeutic directions to inhibit risk factors of PTSD in COVID-19 patients are revealed.
Multiple databases and web portals are used to analyze PPI and pathway enrichments and to identify highly interactive miRNAs, TFs, drugs, chemicals, and diseases.
PSMB9, MX1, and A, C, F, and G of the HLA group were the most unique potential hub proteins.
Several significant GO and metabolic terms, namely adaptive immune system, and IL-18 signaling pathways, were found to be invaded during medical difficulty.
Aberrant expression of significant TFs and miRNAs was found to lead toward comorbidity and cause PTSD in COVID-19 patients.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322241274958 – Supplemental material for Adopting Integrated Bioinformatics and Systems Biology Approaches to Pinpoint the COVID-19 Patients’ Risk Factors That Uplift the Onset of Posttraumatic Stress Disorder
Supplemental material, sj-docx-1-bbi-10.1177_11779322241274958 for Adopting Integrated Bioinformatics and Systems Biology Approaches to Pinpoint the COVID-19 Patients’ Risk Factors That Uplift the Onset of Posttraumatic Stress Disorder by Sabbir Ahmed, Md Arju Hossain, Sadia Afrin Bristy, Md Shahjahan Ali and Md Habibur Rahman in Bioinformatics and Biology Insights
Footnotes
Accessibility to Materials
The corresponding author will be deliberately looking forward to ensuring the availability of the data sets used or investigated, in cases of reasonable requests.
Author Contributions
SaA: Methodology, software, validation, writing - original draft, and data curation; AH: Methodology, software, validation, writing - reviewing & editing; SAB: Writing - reviewing & editing; ShA: Writing - reviewing & editing; HR: Conceptualization, methodology, software, validation, writing - reviewing & editing and supervision.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.