
Abstract
Objectives:
Chagas Disease, caused by the parasite Trypanosoma cruzi, remains a significant public health concern, particularly in Latin America. The current standard treatment for Chagas Disease, benznidazole, is associated with various side effects, necessitating the search for alternative therapeutic options. In this study, we aimed to identify potential therapeutics for Chagas Disease through a comprehensive computational analysis.
Methods:
A library of compounds derived from Cananga odorata was screened using a combination of pharmacophore modeling, structure-based screening, and quantitative structure-activity relationship (QSAR) analysis. The pharmacophore model facilitated the efficient screening of the compound library, while the structure-based screening identified hit compounds with promising inhibitory potential against the target enzyme, sterol-14-alpha demethylase.
Results:
The QSAR model predicted the bioactivity of the hit compounds, revealing one compound to exhibit superior activity compared to benznidazole. Evaluation of the physicochemical, pharmacokinetic, toxicity, and medicinal chemistry properties of the hit compounds indicated their drug-like characteristics, oral bioavailability, ease of synthesis, and reduced toxicity profiles.
Conclusion:
Overall, our findings present a promising avenue for the discovery of novel therapeutics for Chagas Disease. The identified hit compounds possess favorable drug-like properties and demonstrate potent inhibitory effects against the target enzyme. Further in vitro and in vivo studies are warranted to validate their efficacy and safety profiles.
Introduction
Chagas Disease (CD), also known as the American trypanosomiasis, is a neglected tropical disease recognized by the World Health Organization. It is characterized by symptoms such as swelling at the site of infection and fever, and if left untreated, it can lead to congestive heart failure. The disease is caused by exposure to the feces of the triatomine bug, a vector that carries the parasite Trypanosoma cruzi. The name “Kissing Bug” originates from the bug’s tendency to bite people’s faces during feeding. Chagas Disease is endemic to Latin American countries, spanning from Southern United States to Mexico and Argentina. 1 The triatomine bug thrives in poor housing conditions, putting individuals in rural areas at a higher risk of infection. While vector-borne transmission has historically been the primary mode of infection, CD can also be transmitted through blood transfusion and from mother to child during pregnancy. 2
In recent years, significant progress has been made in reducing the transmission of T cruzi through extensive vector control and donor screening programs implemented in many endemic countries. However, several barriers continue to hinder the effective management of the disease. These barriers include challenges related to healthcare financing and payment, limitations in screening and diagnosis methods, suboptimal effectiveness of available treatments, and insufficient awareness among healthcare providers, as well as the general public regarding CD.2,3 Benznidazole and nifurtimox are Food and Drug Administration-approved drugs commonly used to treat CD. However, both drugs have their limitations. Benznidazole, for instance, exhibits a mechanism of action that involves the preferential oxidation of the nucleotide pool, resulting in the incorporation of oxidized nucleotides during DNA replication. This process leads to the formation of potentially lethal double-stranded DNA breaks in the DNA of T cruzi, the parasite causing CD. 4 Despite its effectiveness, benznidazole has certain drawbacks. It is primarily prescribed for children aged 2 to 12 years and has been associated with side effects such as infertility in men and potential harm to unborn babies. 5 In addition, individuals taking benznidazole may experience severe skin reactions, including sore throat, fever, and skin rash. 6 These factors highlight the need for alternative treatment options that can reduce the occurrence of such side effects.
Sterol-14-demethylase is a crucial enzyme belonging to the heme-containing cytochrome P450 family, and it plays a significant role in the synthesis of sterols in eukaryotes.7,8 In protozoa like T cruzi, ergosterol is synthesized as the final product of sterol biosynthesis, contrasting with the production of cholesterol in mammals. The synthesis of ergosterol involves the elimination of the 14α-methyl group from sterol precursors, a process catalyzed by sterol-14-demethylase. 9 In Trypanosomatidae, a family of parasites that includes T cruzi, the causative agent of CD, the synthesis of endogenous sterols is of utmost importance. These sterols not only serve as structural components of the parasites’ membranes but also function as hormonal molecules that regulate various cellular processes, including cell development, multiplication, and morphological transformation, at remarkably low concentrations. 10
While T cruzi can incorporate sterols, primarily cholesterol, from its mammalian host into its membranes, it also relies on de novo sterol synthesis for its survival in all stages of its life cycle. Consequently, the parasite is highly vulnerable to sterol biosynthesis inhibitors. 9 Inhibiting sterol-14-demethylase, the enzyme responsible for a critical step in sterol biosynthesis, has been explored as a potential therapeutic approach against T cruzi and other related parasites.11-13 By targeting this enzyme, it is possible to disrupt the synthesis of essential sterols, which can ultimately lead to the inhibition of parasite growth and survival.
The utilization of plant-derived inhibitors holds great promise in terms of their suitability for oral administration, affordability, and accessibility. One such plant of interest is Cananga odorata, commonly known as “ylang-ylang,” which has a rich history as a traditional medicinal plant in South-Eastern Asian countries like the Philippines and Malaysia. 14 C odorata is a medium-sized tree that can reach heights of up to 15 m. Extensive research has highlighted the diverse therapeutic properties of C odorata. It has been recognized for its antimicrobial, antibiofilm, antioxidant, insecticidal, anti-inflammatory, and antibacterial effects.14,15 In a study, three compounds extracted from C odorata, namely O-methylmoschatoline, liriodenine, and 3,4-dihydroxybenzoic acid, exhibited notable antibacterial and antifungal activities. 16 Furthermore, another constituent from C odorata, N-trans-feruloyltyramine, identified in the methanolic extract of the seeds, was found to potentially contribute to the suppression of melanogenesis by inhibiting the expression of the tyrosinase enzyme. 17
These bioactive properties make C odorata a valuable resource for potential drug discovery and development. By harnessing the beneficial compounds present in C odorata, it may be possible to develop novel oral treatments that are cost effective and readily available.
This study utilized computational techniques to identify potential inhibitors of sterol-14-demethylase from C odorata. The objective was to find phytoconstituents with anti-trypanosomal activity. The workflow included structure-based screening, molecular docking, quantitative structure-activity relationship (QSAR) analysis, pharmacophore modeling, and pharmacokinetic assessments. The findings could contribute to the development of novel drugs for treating trypanosomal infections using compounds from C odorata.
Materials and Method
Protein selection and preparation
The sterol-14-alpha demethylase enzyme, with a Protein Data Bank (PDB) ID of 4CKA, was obtained from the official website of the PDB (https://www.rcsb.org/). Before its use in this study, the protein underwent thorough preprocessing and was subjected to modifications aimed at optimizing the assignment of hydrogen bonds and minimizing its structure using the OPLS3e force field. 18 The resulting minimized protein was then utilized for further analyses and investigations.
Ligand generation and preparation
A compound library for C odorata was generated in SDF format by retrieving relevant compounds from the PubChem web database (Supplemental Table 1). The retrieved compounds underwent further processing using the LigPrep tool. This tool facilitated the conversion of the initially available two-dimensional molecules into three-dimensional structures. The process involved ionizing the molecules at a pH of 7.2 ± 0.2 and eliminating any salts present using Epik. The OPLS3 force field was applied to ensure accurate ionization and tautomeric state representation of the compounds. 19
Receptor grid generation
To identify the specific region of interaction between the ligand and protein, a receptor grid was created. This grid, known as the Glide Grid, was constructed using the Receptor Grid Generation tool and focused on the binding domain of the protein. 20 By selecting the co-crystallized ligand situated at the active site of 4CKA, the binding location was determined. To facilitate the subsequent process of molecular docking, a grid box was automatically generated for the protein. The dimensions of the grid box were defined as follows: X = 0.86, Y = 27.49, and Z = 456.75.
Pharmacophore modeling and screening
The receptor-ligand complex was extensively studied, and a hypothesis (E-pharmacophore) was generated using the phase interface within the Schrödinger suite. This pharmacophore was designed to highlight the key properties that play a crucial role in the specific binding of the ligand to the active sites of the target protein. 21 Subsequently, the hypothesis was employed as a filtering criterion to eliminate compounds that did not exhibit at least two out of the five essential features identified by the pharmacophore analysis.
Molecular docking
The molecular docking process was performed using the Glide tool, utilizing Maestro 11.1 software. The compound library, which had already been filtered down to 28 compounds, was docked into the prepared grid of protein targets. Two docking algorithms were employed: Standard Precision (SP) and the more rigorous Extra Precision (XP) algorithms. Initially, compounds with a docking score of less than −5.0 kcal/mol using the SP algorithm were filtered out, resulting in the exclusion of four compounds. The remaining selected compounds (24) were then subjected to XP docking. During the docking process, the protein was considered a rigid body, while the ligand’s rotatable bonds were allowed to freely move and adopt different conformations. 22
Molecular mechanics grown born surface area
Molecular mechanics-generalized Born surface area (MM-GBSA) is a computational method used to calculate the energy of various components involved in a molecular system, including optimized free receptors, free ligands, and the complex formed by the ligand binding to the receptor. In addition, it enables the evaluation of ligand strain energy by placing the ligand in a solvent environment generated by the VSGB 2.0 suite. In this study, Prime rotamer search techniques available in Maestro were employed in conjunction with the OPLS3 force field and the VSGB solvent model. These tools and models were utilized to perform the necessary calculations and simulations. The binding free energy, which quantifies the strength of the interaction between the ligand and receptor, was determined using the following equation:

This equation takes into account various energy contributions from the molecular system and provides valuable insights into the binding affinity between the ligand and receptor. 21
Development of AutoQSAR Model
To gather information about sterol-14-alpha demethylase inhibitors’ activity, a total of 38 inhibitors were retrieved from the CHEMBL database. The protein’s FASTA sequence, obtained from the PDB, was used as a query to extract these compounds. To facilitate further analysis, the extracted compounds were converted into the “.SDF” file format using Data Warrior software. 18 Using the AutoQSAR module, a QSAR model was constructed utilizing these compounds. Multiple models were generated, and among them, the kpls_radial_44 model was identified as the best-performing model based on its rank and predictive capability. Subsequently, this selected model, kpls_radial_44, was employed to predict the pIC50 values of the top-ranked compounds as well as a standard compound. This QSAR model allowed for the estimation of the inhibitory activity of these compounds, providing valuable insights into their potential as sterol-14-alpha demethylase inhibitors.
Absorption Distribution Metabolism Excretion/Toxicity screening
To assess the absorption, distribution, metabolism, and excretion (ADME) characteristics of the top five compounds, as well as the standard drug, their efficacy was predicted using the admetSAR server. The admetSAR server (http://lmmd.ecust.edu.cn/admetsar2/) provides valuable predictions regarding the ADME properties of compounds. These properties play a vital role in drug development as they help evaluate the potential of a compound to become an effective drug. By analyzing these characteristics, valuable insights can be gained regarding the compound’s ADME, contributing to the decision-making process in drug discovery and development. 22
Result and Discussion
Before molecular docking, a pharmacophore hypothesis was developed based on the complex formed by the protein and co-crystallized ligand. The PHASE module in Schrödinger suite was employed to extract essential information regarding the molecular orientation of crucial functional groups involved in the high-affinity binding of ligands to the protein target.
The resulting E-pharmacophore hypothesis (Figure 1) consisted of two hydrophobic groups and three aromatic rings. The pharmacophore hypothesis was used to filter the library of compounds, allowing for the identification of ligands that possess similar key features important for binding to the target protein.

Pharmacophore hypothesis generated.
The molecular docking analysis identified five compounds from C odorata that demonstrated strong inhibitory potential against the target enzyme, with docking scores comparable to the standard compound benznidazole (Supplemental Figure 1). A more negative docking score indicates a higher inhibitory potential. The compounds benzyl salicylate, calamenene, benzyl benzoate, 5-indanol, and p-cymene exhibited excellent inhibitory potential, with docking scores of −7.627, –7.453, –6.305, –6.151, and −6.058 kcal/mol, respectively, compared to the docking score of −6.817 kcal/mol for benznidazole, which is a standard drug used to treat CD (Table 1).
Docking scores of the top-scoring compounds and standard against sterol-14-alpha demethylase.

Abbreviation: MM-GBSA, molecular mechanics-generalized Born surface area.
Structural-based drug design focuses primarily on the interaction between the protein and ligand, as the extent of inhibition is largely determined by the ligand’s interaction with specific amino acid residues at the active site of the target enzyme. 23 Table 2 and Figure 2 present the key interactions contributing significantly to the inhibition of sterol-14-demethylase in this study. Benzyl salicylate formed a single hydrogen bond with LYS 421, while calamenene formed a single PI-PI stacking bond with TYR 103. Benzyl benzoate also formed a single PI-PI stacking bond with PHE 290. 5-Indanol made a single H-bond interaction with ALA 287. Benznidazole, the standard, made two H-bonds with TYR 116.

Two-dimensional interaction of the hits and the standard. Benznidazole in the active site of the enzyme. (A) Benzyl salicylate, (B) calamenene, (C) benzyl benzoate, (D) 5-indanol, (E) p-cymene, (F) benznidazole (standard).
Interaction profile of top-ranked compounds including the standard within the active site of sterol-14-alpha demethylase.

In addition, the MM-GBSA technique provides a more accurate estimation of the binding free energies (dG) for protein-ligand complexes. Negative values indicate stable complexes in the target’s binding pocket. In this study, all lead compounds exhibit negative values, indicating their stability within the target’s binding pocket. The binding free energies for the docked complexes were −;43.729, –30.424, –38.262, –31.948, and −31.003 kcal/mol for benzyl salicylate, calamenene, benzyl benzoate, 5-indanol, and p-cymene, respectively. This suggests that all the hit compounds are more stable in the target’s binding pocket. Benznidazole, the standard, exhibits a binding free energy of −41.157 kcal/mol. The utilization of the MM-GBSA technique enhances the accuracy of virtual screening results and provides valuable insights into the stability of protein-ligand complexes within the binding pocket. The negative binding free energy values obtained for the lead compounds indicate their favorable binding characteristics, further supporting their potential as effective inhibitors.
AutoQSAR
Quantitative structure-activity relationship analysis is a crucial computational tool in drug discovery that explores the relationship between the structural characteristics of small molecules and their biological activities. 24
In this study, the AutoQSAR module within the Schrodinger suite was employed, utilizing various topological descriptors to construct independent variable models based on experimental data generated for the target.
Among the generated QSAR models, the kpls_radial_44 model (Figure 3) emerged as the best-performing model, trained using machine learning techniques. The model’s performance was evaluated using key parameters such as SD (0.4412), coefficient of determination (0.8581), root mean squared error (0.695), and cross-validation (0.6087; Table 3). Table 4 shows the test and training set utilized in generating the model.
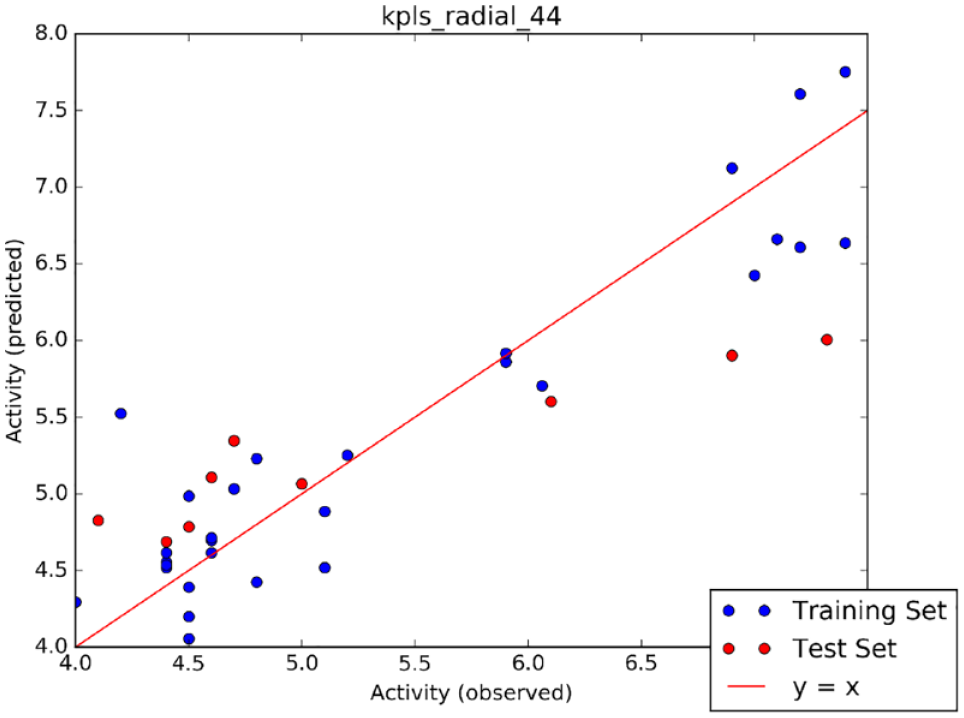
Scatter plot for the best AutoQSAR model (kpls_radial_44).
Parameters of the QSAR model.
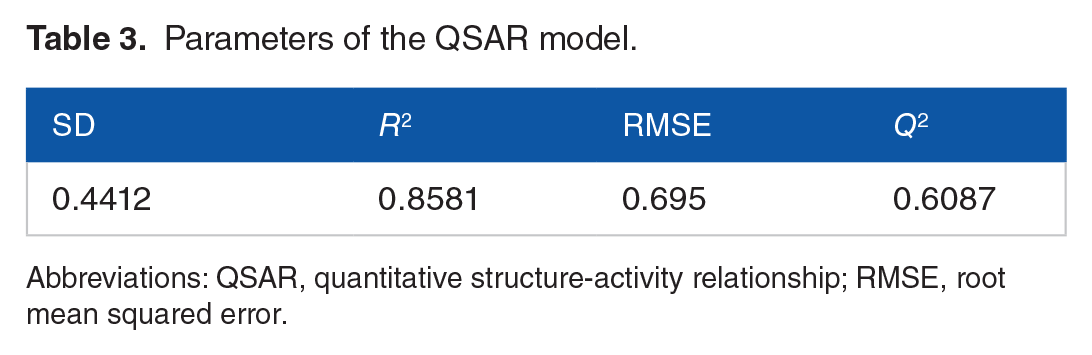
Abbreviations: QSAR, quantitative structure-activity relationship; RMSE, root mean squared error.
Train and test set for the QSAR model.

Abbreviation: QSAR, quantitative structure-activity relationship.
Subsequently, the QSAR model was employed to predict the pIC50 values of the hit compounds listed in Table 5. The obtained pIC50 values for the hit compounds are also presented in Table 5. Notably, only benzyl benzoate demonstrated a superior pIC50 value (5.349 nM) compared to the standard drug (5.331 nM).
Predicted pIC50 values of hit compounds and standard.

ADMET and Drug Likeness
The analysis of ADME is a crucial aspect of the drug discovery process, providing valuable insights into the behavior of drugs within a biological system. 25 In this study, we employed the admetSAR server to predict the drug-like properties and potential toxicity of the selected lead compounds and the standard compound.
To evaluate the drug likeness of the compounds, we employed the Lipinski rule, which encompasses five key criteria for determining oral activity. 26 The results, presented in Table 6, demonstrate that all of the selected compounds exhibit favorable physicochemical properties, including low molecular weight, a desirable bioavailability score, and good solubility. Notably, these properties position the lead compounds as top drug-like candidates based on their physicochemical profiles. In addition, none of the compounds violated more than one of the Lipinski rule criteria. The implicit log P (iLogP) value is utilized to measure the n-octanol/water partition coefficient of compounds, measuring their lipophilicity. Lipophilicity impacts a drug’s solubility and permeability across membranes, with iLogP being a predictive method used for this purpose. 27 All compounds, including the standard, were predicted to exhibit lipophilic characteristics, as their values were below five. Veber’s rule says that compounds with a polar surface area (TPSA) <140 Å 2 tend to have favorable oral bioavailability. In this case, all compounds displayed TPSA scores below 140. The logKp values, measured in cm/s, for the compounds, including the standard, were predicted to range from −3.96 to −7.24. A more negative logKp value suggests a lower ability for the molecule to pass through the skin. 28 Benznidazole, the standard drug, was predicted to have the lowest skin permeability score (–7.24 cm/s).
Drug likeness prediction of the hit compounds and standard.
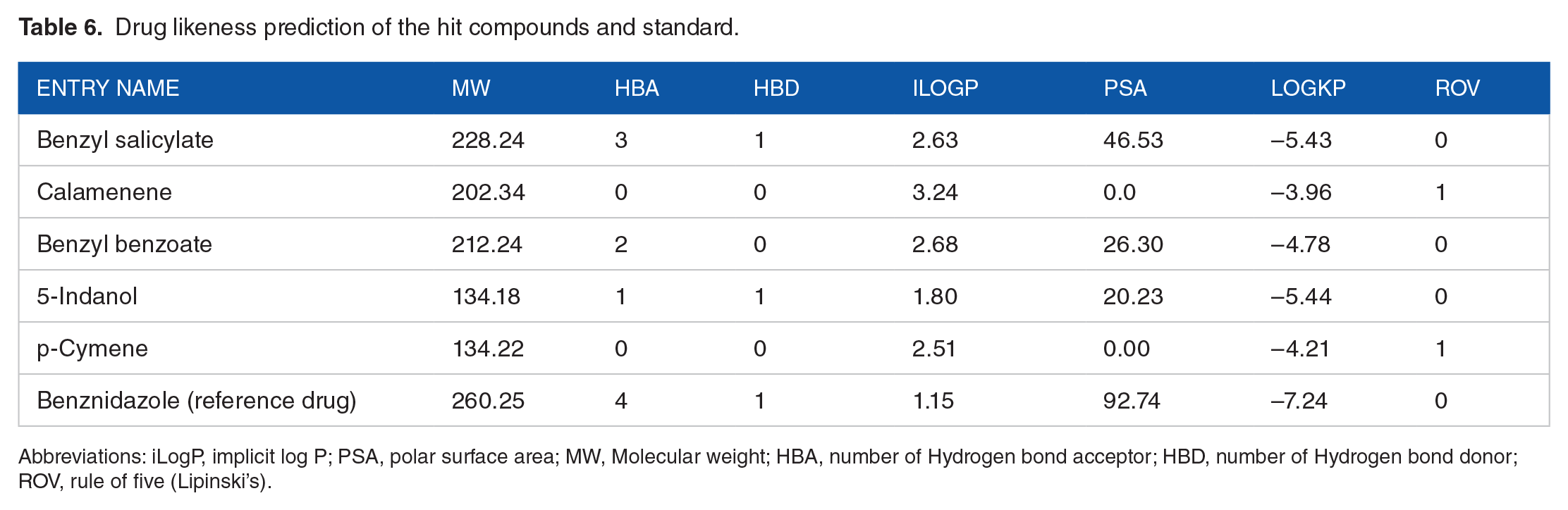
Abbreviations: iLogP, implicit log P; PSA, polar surface area; MW, Molecular weight; HBA, number of Hydrogen bond acceptor; HBD, number of Hydrogen bond donor; ROV, rule of five (Lipinski’s).
In terms of blood-brain barrier (BBB) permeability, all test compounds, except benzyl salicylate, were predicted to cross the BBB. The BBB serves as a crucial barrier that regulates the entry of molecules into the central nervous system from the bloodstream, controlling access to the brain. 29 P-glycoprotein (P-gp), a transmembrane efflux pump, plays a role in expelling drugs out of cells, which can potentially lead to poor drug efficacy or therapeutic failure. 30 However, the results indicate that the hit compounds, including the standard, are non-substrates and non-inhibitors of P-gp. This suggests that these compounds are less likely to be affected by P-gp-mediated efflux and may exhibit improved drug effectiveness.
The cytochrome P450 (CYP450) enzymes are responsible for metabolizing therapeutic drugs and play a crucial role in drug clearance within the liver. 31 Inhibition of CYP450 isoforms can lead to drug-drug interactions and impact drug toxicity profiles. In our assessment, we found that none of the hit compounds, including the standard, were predicted to be substrates or inhibitors of CYP3A4, a commonly studied CYP450 isoform. Similar results were observed for the CYP2C9 isoform, indicating a lower likelihood of drug interactions mediated by these enzymes. Furthermore, none of the compounds were predicted to inhibit CYP2D6 (Figure 4).
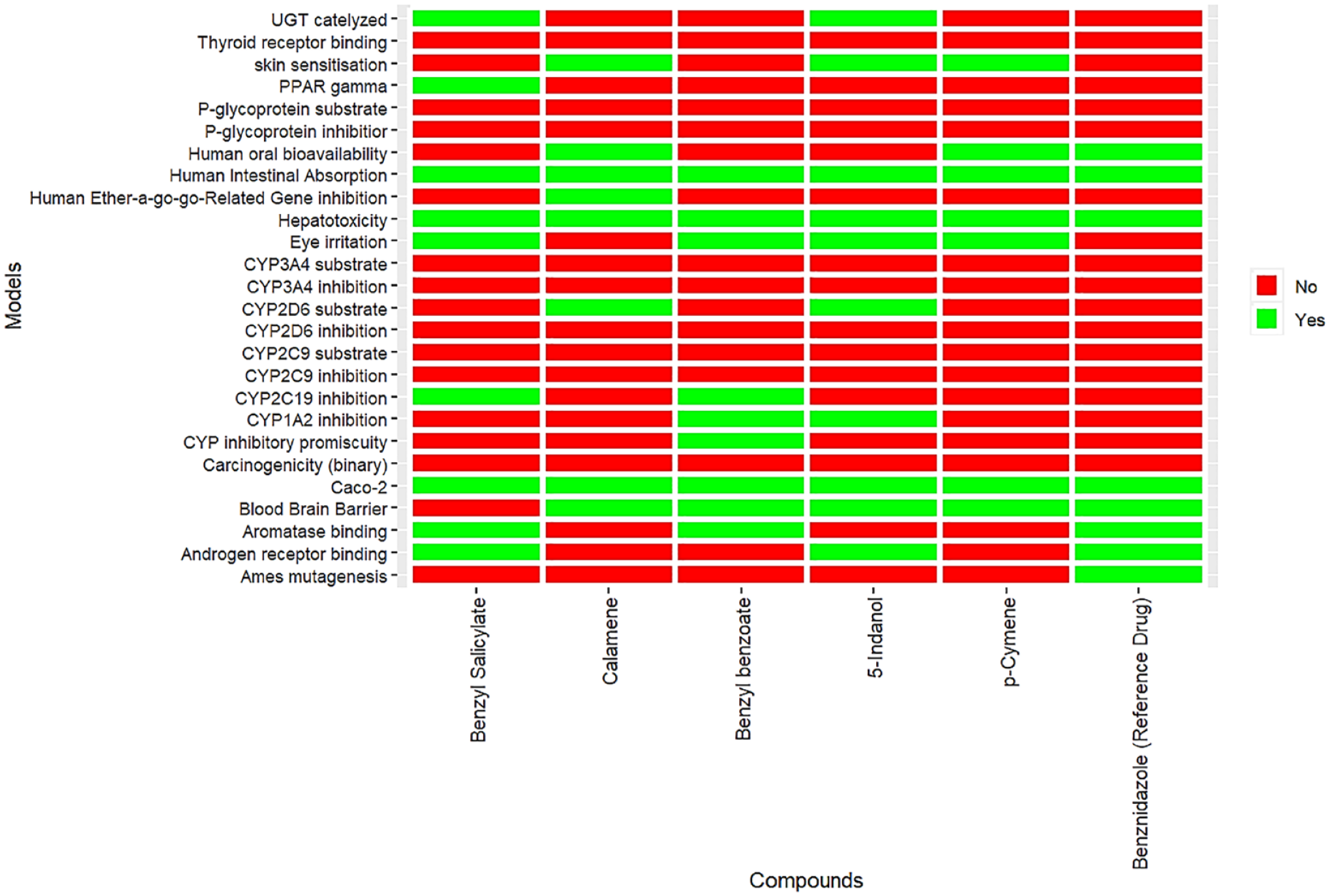
ADMET properties of the hit compounds and standard.
Toxicity analysis revealed that all compounds, including the standard drug, demonstrated non-carcinogenic and non-eye corrosive properties. However, it is important to note that only the standard drug was predicted to have mutagenic potential based on the results of the Ames mutagenesis test, which assesses the likelihood of a chemical causing mutagenic or carcinogenic effects. 32
Various studies have explored the inhibition of sterol-14-demethylase through different approaches. Kulactone and gallocatechin emerged as potent inhibitors in an in silico study, as determined by extensive molecular dynamics simulations. 22 Alternatively, a study focused on synthesizing potential antimicrobial compounds, specifically 1,3-phenylene-based symmetrical bis(urea-1,2,3-triazole) hybrids, demonstrated inhibitory effects on the sterol-14-demethylase enzyme. Benzyl salicylate, identified as a hit compound in this study, demonstrated anti-inflammatory activity by inhibiting the expression of inducible nitric oxide synthase and cyclooxygenase-2. 33 In addition, calamenene (also identified as a hit compound in this study) and its analogs, Cala 1 and Cala 2, reported in another study, exhibited antibacterial properties. 34
Conclusion
In conclusion, this study focused on identifying potential therapeutics for CD through a comparative analysis with the standard drug, benznidazole. I employed a computational approach, utilizing various tools and models to screen a library of compounds derived from C odorata. Through pharmacophore modeling and structure-based screening, I successfully identified several hit compounds that showed promising bioactivity against the target enzyme, sterol-14-alpha demethylase. In addition, the QSAR model predicted one of the hits to exhibit superior bioactivity compared to benznidazole. Furthermore, I conducted a comprehensive evaluation of the physicochemical, pharmacokinetic, toxicity, and medicinal chemistry properties of the identified lead compounds. The results indicated that these compounds possess drug-like characteristics, are orally bioavailable, are easily synthesizable, and exhibit reduced toxicity profiles, making them potential candidates for further development.
While these findings provide valuable insights and initial evidence of the efficacy of these compounds, further in vitro and in vivo studies are necessary to confirm their therapeutic potential. Continued research and evaluation of these compounds will contribute to the development of alternative and safer treatments for CD, addressing the limitations associated with the current standard therapy.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322241262635 – Supplemental material for Machine Learning-Based Approach to Identify Inhibitors of Sterol-14-Alpha Demethylase: A Study on Chagas Disease
Supplemental material, sj-docx-1-bbi-10.1177_11779322241262635 for Machine Learning-Based Approach to Identify Inhibitors of Sterol-14-Alpha Demethylase: A Study on Chagas Disease by Jamiyu A Saliu in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
None
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Jamiyu A Saliu performed the study and wrote the manuscript.
Ethical Statement
Not applicable
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.