
Abstract
The ecological deterioration caused by the continuous and excessive use of synthetic inputs in agriculture has prompted the search for environmentally favorable resources for crop production. Many have advocated for the use of soils from termite mounds to improve soil and plant health; therefore, the purpose of this study was to characterize the microbiome multifunctionalities that are important for plant health and growth in termite mound soil. The metagenomics of soil from termite mounds revealed taxonomic groups with functional potentials associated with promoting the growth and health of plants in nutrient-poor, virtually dry environments. Analysis of microorganisms revealed that Proteobacteria dominated the soil of termite colonies, while Actinobacteria ranked second. The predominance of Proteobacteria and Actinobacteria, the well-known antibiotic-producing populations, indicates that the termite mound soil microbiome possesses metabolic resistance to biotic stresses. Functions recognized for diverse proteins and genes unveiled that a multi-functional microbiome carry out numerous metabolic functions including virulence, disease, defense, aromatic compound and iron metabolism, secondary metabolite synthesis, and stress response. The abundance of genes in termite mound soils associated with these prominent functions could unquestionably validate the enhancement of plants in abiotic and biotically stressed environments. This study reveals opportunities to revisit the multifunctionalities of termite mound soils in order to establish a connection between taxonomic diversity, targeted functions, and genes that could improve plant yield and health in unfavorable soil conditions.
Introduction
Termites are highly diverse and are mainly found in Africa, Asia, and other continents except Antarctica. 1 There are over 2 600 well-known termite species, and approximately 20% of them have detrimental effects on agricultural produce. 2 Through the excavation of materials from parent soils, mound-building termites contribute significantly to ecological services when building their mounds. 3 Termites play significant roles in enhancing the physical and chemical properties of soil (eg, nitrogen, phosphorus, potassium, clay) and breaking down organic matter into forms utilizable by plants. 4 Termites as “soil engineers” influence the distribution of nutrients and minerals to their adjacent soil. 5 Termites construct structures called mounds which have been reviewed by several authors to be rich in microbial diversity as influenced by a lot of nutrients accumulated in soil from termite mound.6,7
Recently, soils from termite mound have been presented to farmers in central Côte d’Ivoire, 8 Sierra Leone, 9 Zimbabwe 10 , and Uganda 11 to grow vegetables and other crops on top of mounds, whereas agronomists in southern Zambia excavate and amass soil from termite mounds and use it as an alternative to increase their soil fertility at their local farms. 12 This idea will reduce the total reliance on chemical fertilizers or pesticides by farmers and then reduce the negative impact of excessive and prolonged use of chemical fertilizers on the environment. 13 In accordance with this recommendation, we chose to investigate the effects of termite bioturbation on the termite mound soil microbiome and determine their role in promoting plant development, health, and disease resistance through adaptation to abiotic and biotic stresses, xenobiotic degradation, stress responses, and iron acquisition. This research will assist in understanding how diverse microorganism communities in termite mound soils may impact plant performance and productivity (as reported by local subsistence farmers).14,15 To achieve this aim and surmount the limitations of the culture-dependent approach, a shotgun sequencing approach was used to completely characterize the microbial communities and functional genes in termite mound soils.16,17 This metagenomic approach may unleash novel opportunities for developing environmentally friendly techniques to generate profits from microbe-assisted farming machinery. With this in mind, it was hypothesized that soil from termite mounds contains a large gene pool for (a) detoxification of xenobiotic compounds and metals, (b) resistance against diverse antimicrobial compounds, (c) iron-acquisition functions that enhance iron bioavailability, and (d) numerous secondary metabolite pathways associated with the production of antibacterial peptides or bacteriocins.
Materials and Methods
Study sites and soil sampling
This research was done in agricultural areas in a nearly dry area of South Africa’s North West province (25°27’11.2”S 26°07’33.8”E; 25°26’13.5”S 26°05’50.4”E). This province shares border with Botswana and is defined by trees, shrubs, and background distribution of mountains in the north-east. The average winter temperature ranges from 3°C to 21°C, and the average summer temperature ranges from 17°C to 31°C. The average annual rainfall is 360 mm, with the heaviest precipitation occurring in the tenth and fourth month of the year. 18 For this investigation, 50 g of soil was collected from termite mounds ranging in depth from 0 to 15 cm at the base of termite mounds where the Coptotermes species construct nests resembling boxes and where their bioturbation had effect. 19 Eight termite mound soil samples (in each termite mound, we collected three replicates, which were pooled as one before analysis) from the agricultural areas (T1-T8) were collected using a split tube auger 5 m in diameter. The mounds were populated by Coptotermes sp as identified by a method previously used by Arif et al. 20 The soil samples were stored in coolers for the period of sample collection and shipment to the laboratory, and it arrived at the laboratory on the day of collection for analysis. The soil samples were deposited for 2 weeks in the refrigerator (at 4°C) before DNA extraction and the determination of physicochemical properties.
Assessment of soil physical and chemical properties
The soil physicochemical components were evaluated within 14 days of collection. Twenty grams of soil was desiccated, grounded, and sifted (2-mm sieve) to remove unwanted particles. The particle size was determined using hydrometer techniques, and textural classifications were assigned accordingly. Textural classes were determined in accordance with the US Department of Agriculture’s regulations: The particulate sizes of sand, silt, and clay range between 2.0 mm and 0.05 mm, 0.05 mm and 0.002 mm, and less than 0.002 mm, respectively. The soil was mixed with distilled water (1:2.5), and pH was evaluated using a pH meter. The total nitrogen values were obtained using the Kjeldahl process as illustrated by Muwawa et al. 21 Potassium (K), magnesium (Mg), and calcium (Ca) were examined after mining using 1 mol dm−3 NH4CH3CO2 (ammonium acetate) at a pH of 7.0. An atomic absorption spectrophotometer was used to read the exchangeable Ca and Mg, and the exchangeable K was measured with a flame photometer. 22 Spectrophotometer was used to measure the available phosphorus (P), whereas dichromate digestion was used to measure the organic carbon available in the soil samples. 23
DNA extraction and sequencing
With the aid of the PowerSoil DNA isolation kit (MoBio Laboratories, Inc., Carlsbad, CA, USA), we extracted the full genomic DNA from 0.25 g of each soil sample, by adhering to the company’s manual. The whole data were generated by shotgun metagenome sequencing at the MR DNA Lab, Shallowater, Texas. The DNA concentrations were evaluated by fluorescence using the Quant-iT PicoGreen dsDNA kit (Invitrogen, Carlsbad, CA, USA) and a DQ 300 fluorometer (Hoefer Scientific Instruments, California). Libraries were built using 50 ng of DNA from individual specimens via the Illumina Nextera DNA Sample Preparation Kit (San Diego, CA, USA). An Experion automated electrophoresis station (Bio-Rad Laboratories, 3, Boulevard Raymond Poincaré 92430 Marnes-la-Coquette, France) was used to decide the library insert size, which varied from 300 to 850 bp (average 500 bp). Individually, the library was put into an Illumina 600-cycle v3 reagent container and then sequenced using 2 × 250 bp sequencing carried out on the Illumina MiSeq.
Metagenome annotation analyses
Individual unprocessed metagenome sequences were evaluated using a widely accessible online pipeline called MG-RAST (which stands for Metagenomic Rapid Annotations using Subsystems Technology). The raw sequences were subjected to quality control to eliminate false sequences, host-specific sequences, ambiguous base pairs, and sequences with a length greater than two standard deviations above the mean. Annotation of sequences was performed by blasting against the M5NR database infrastructure, which offers nonredundant incorporation of numerous databases. 24 For the taxonomic groupings, sequence alignment was performed via the MG-RAST pipeline against the RefSeq protein database. The nonsupervised orthologous group (NOG), clusters of orthologous groups (COG), Kyoto Encyclopedia of Genes and Genomes (KEGG), and SEED Subsystem were used for the functional annotation of diverse metabolic pathways/genes under the following conditions: An e-value of 0.00001, a least identity of 60%, and an utmost alignment length of 15 bp were the parameters utilized for the assignment of taxonomic assignments. 25 Sequences obtained from viruses and eukaryotes (except fungi) were rejected for this study. To reduce the effect of experimental errors, the standardized choice of MG-RAST was used. The microbiome tables obtained afterward were classified to their individual taxa levels for statistical purposes to obtain their rank abundance at the phylum level. Subsequently, the abundances were converted into percentages. We deposited the sequences utilized in this research in the Sequence Read Archive of the National Center for Biotechnology Information under the bioproject number PRJNA526912. Shinyheatmap was used for the generation of heatmaps; the z-scores were transformed to the relative abundances of microbiome. 26
Results
Physicochemical characterization of soil from termite mounds
Physicochemical analysis of termite mound soil varied among samples (Supplemental Table S1). However, termite mound soil was rich in soil nutrients like nitrogen, organic carbon, Ca, K, and Mg.
Overall assessment of sequencing data of the termite mound soils
The sequences uploaded were from 6 949 161 to 6 984 205 reads for the soil samples, whereas the left-over sequences after preprocessing were from 6 422 685 to 6 802 220, with a mean G + C value of 61.25%.
Microbiome composition and abundance
An aggregate of 37 diverse phyla (related to bacteria, archaea, and fungi) were found for the reads. Among all the phyla, Proteobacteria (between 34.27% and 53.48%) and Actinobacteria (between 30.11% and 43.56%) were the predominant communities found in the samples. Some phyla from archaea and fungi were also present (Figure 1).
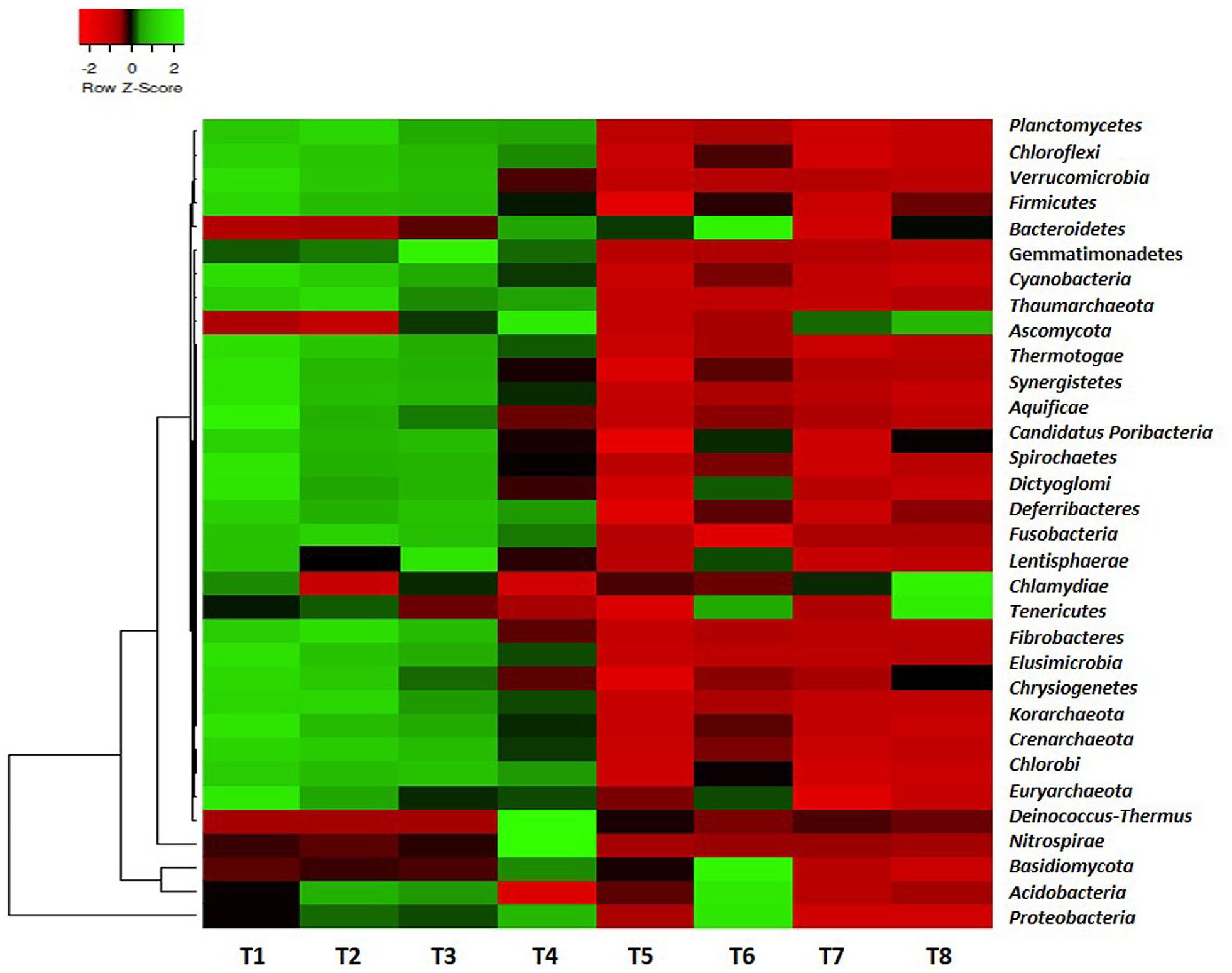
The rank abundance of the microbiome in the termite mound soils.
Metabolic multifunctionalities in the termite mound soil
Protein function in the sequence data was characterized with KEGG, NOG, COG, and SEED Subsystem databases, evidence-based annotations. KEGG classification indicated that metabolism (61.30%) was the predominant group, followed by genetic information processing (17.69%), environmental information processing (15.39%), cellular processes (3.85%), human diseases (1.46%), and organismal systems (0.32%) (Figure 2). NOG’s cataloging revealed that most of the protein functions in the data set were poorly characterized (76.76%). However, metabolism (10.61%), information storage and processing (7.40%), and cellular processes and signaling (5.23%) were recorded (Figure 2). COG’s taxonomy revealed that the relative abundances of metabolism, cellular processes and signaling, and information storage and processing were 46.44%, 19.03%, and 17.09%, respectively. However, a poorly characterized category (17.44%) was also noticed (Figure 2). Ranked examination of SEED Subsystems (level 1) revealed diverse key microbial functions in the sequence data set as reported in Supplemental Table S2.

Reads to different functional groups depicted by NOG, KEGG, and COG databases, while a subsystem database has been reported in Supplemental Table S2.
Secondary metabolism
Biological pathways and processes that were obviously identified belonged to the nonribosomal peptide synthetases (NRPS) in Frankia sp. Ccl3, flavanone biosynthesis, alkaloid biosynthesis from L-lysine, auxin biosynthesis and degradation, phenazine biosynthesis, steroid sulfates, tannin biosynthesis, and cinnamic acid degradation. Furthermore, quinolinic acid and its derivatives, clavulanic acid biosynthesis, paerucumarin biosynthesis, phenylpropionate degradation, 2-isocapryloyl-3R-hydroxymethyl-gamma-butyrolactone, steroid sulfates, octadecanoids, pyrrolnitrin biosynthesis, cannabinoid biosynthesis, phytoalexin, phytosterol, salicylic acid, suberin, lignin biosynthesis, and phenylpropanoids general biosynthesis were also identified in mound soils (Figure 3). We also identified genes such as 2,3-dihydroxyphenylpropionate 1,2-dioxygenase, aromatic-L-amino-acid decarboxylase (EC 4.1.1.28), tryptophan synthase alpha chain (EC 4.2.1.20), nitrilase 1 (EC 3.5.5.1), aromatic-L-amino-acid decarboxylase (EC 4.1.1.28), indole-3-pyruvate decarboxylase (EC 4.1.1.74), nitrilase 2 (EC 3.5.5.1), Hca operon (3-phenylpropionic acid catabolism) transcriptional activator HcaR, and probable 3-phenylpropionic acid transporter (Supplemental Table S3).
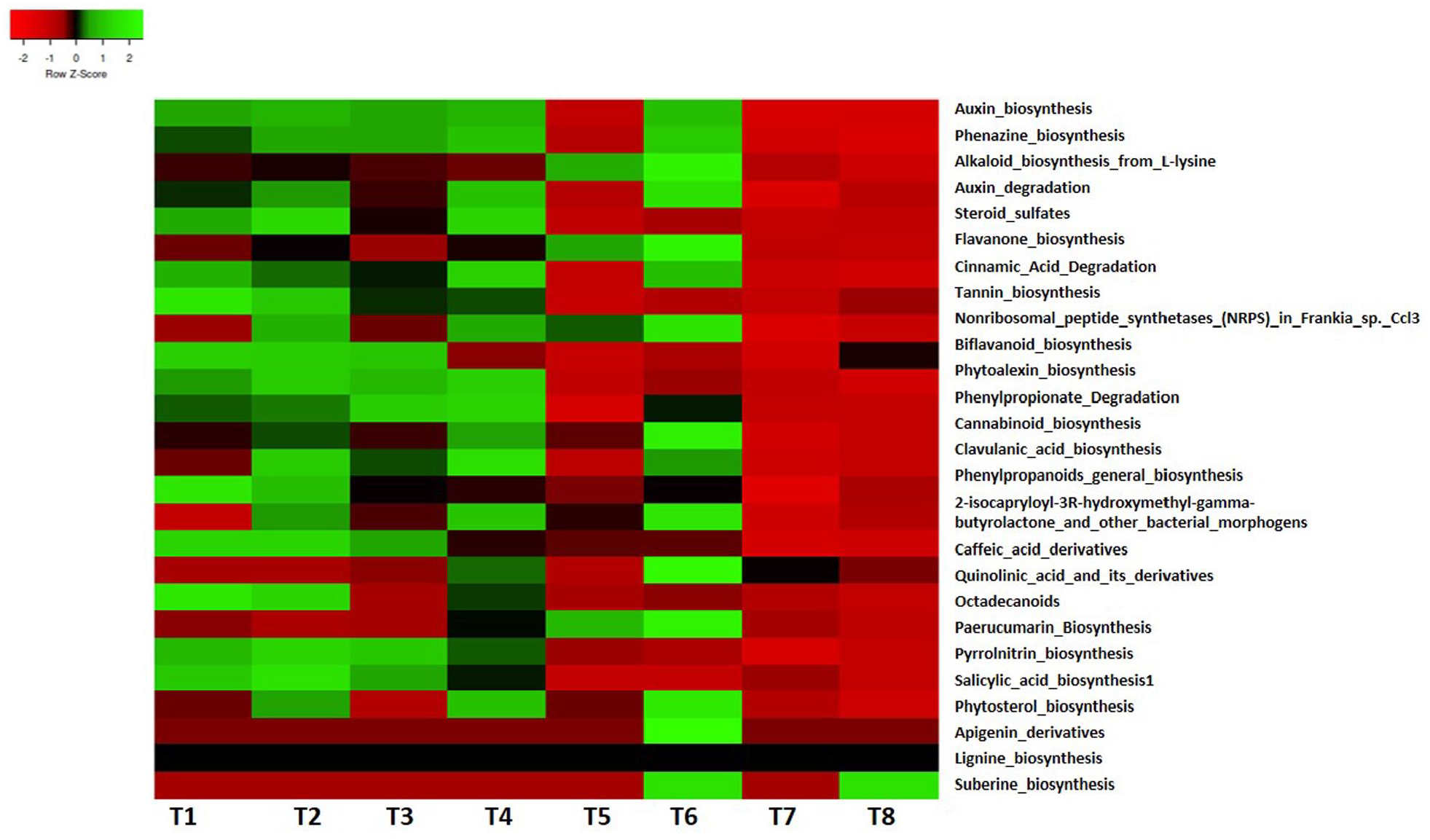
Pathways linked to secondary metabolism in termite mound soils.
Metabolism of aromatic compounds
Sequence reads associated with several pathways involved in the peripheral pathways for catabolism of aromatic molecules, metabolism of aromatic intermediates, aromatic compounds, and anaerobic degradation of xenobiotic compounds were recognized in this study (Figure 4). Metabolic pathways that predominated the termite mound soils include n-phenylalkanoic acid, benzoate transport and degradation cluster, anaerobic benzoate metabolism, phenylpropanoid compound degradation, N-heterocyclic aromatic compound degradation, protocatechuate branch of the beta-ketoadipate pathway, catechol branch of the beta-ketoadipate pathway, phenylacetyl-coenzyme A (CoA) catabolic pathway (core), and homogentisate pathway of aromatic compound degradation. Also observed in termite mound soils are genes involved in the degradation of naphthalene and anthracene (like naphthalene dioxygenase ferredoxin and naphthalene 1,2-dioxygenase system ferredoxin—NAD[+] reductase component [EC 1.18.1.3], 1,2-dihydroxynaphthalene dioxygenase), central meta-cleavage of aromatic compounds, 4-hydroxyphenylacetic acid catabolic pathway (like 4-hydroxyphenylacetate 3-monooxygenase [EC 1.14.13.3], 3,4-dihydroxyphenylacetate 2,3-dioxygenase [EC 1.13.11.15], and 4-hydroxyphenylacetate 3-monooxygenase, reductase component [EC 1.6.8.-]), quinate (like 3-dehydroshikimate dehydratase, protein QA-X, and quinate permease), anaerobic toluene and ethylbenzene (like [S]-1-phenylethanol dehydrogenase, (R)-benzylsuccinyl-CoA dehydrogenase [EC 1.3.99.21], and acetophenone carboxylase subunit Apc2), p-hydroxybenzoate, pathways for degradation of gentisare (like fumarylacetoacetate hydrolase family protein, gentisate 1,2-dioxygenase [EC 1.13.11.4], and putative 4-hydroxybenzoyl-CoA thioesterase), chlorobenzoate (like 2-chlorobenzoate 1,2-dioxygenase alpha subunit [EC 1.14.12.13], 2-chlorobenzoate 1,2-dioxygenase beta subunit [EC 1.14.12.13], and 2-chlorobenzoate 1,2-dioxygenase reductase component), benzoate (like 1,2-dihydroxycyclohexa-3,5-diene-1-carboxylate dehydrogenase [EC 1.3.1.25], 2-chlorobenzoate 1,2-dioxygenase alpha subunit [EC 1.14.12.13], and 2-chlorobenzoate 1,2-dioxygenase beta subunit [EC 1.14.12.13]), biphenyl (like 2,3-dihydroxybiphenyl 1,2-dioxygenase, 2-hydroxypenta-2,4-dienoate hydratase, and 2-keto-4-pentenoate hydratase [EC 4.2.1.-]), chloroaromatic (like chlorocatechol 1,2-dioxygenase), salicylate ester, and toluene (like 1,2-dioxygenase alpha subunit, toluate 1,2-dioxygenase electron transfer component, and toluene-4-monooxygenase, subunit TmoD) (Figure 4, Supplemental Table S4).

Pathways linked to the metabolism of aromatics in termite mound soils.
Stress response
The prominent metabolic pathways linked with stress response in termite mound soils were regulation of oxidative stress response, oxidative stress, choline and betaine uptake and betaine biosynthesis, glutathione biosynthesis and gamma glutamyl cycle, synthesis of osmoregulated periplasmic glucans, sigmaB stress response regulation, bacterial hemoglobins, uptake of selenite, protection from reactive oxygen species, glutathione nonredox reactions, and heat shock DnaK gene cluster extended (Figure 5). Other stress response paths identified from the termite soils include periplasmic stress response, cold shock CspA family of proteins, carbon starvation, acid resistance mechanisms, and osmotic stress cluster. Genes linked to the enzymes of the pathways involved in osmoregulation (like aquaporin Z, osmotically inducible protein OsmY, and propanediol diffusion facilitator); ectoine biosynthesis and regulation (like diaminobutyrate-pyruvate aminotransferase [EC 2.6.1.46], L-2,4-diaminobutyric acid acetyltransferase [EC 2.3.1.-], and L-ectoine synthase [EC 4.2.1.-]); osmoprotectant adenosine triphosphate (ATP)-binding cassette (ABC) transporter (osmoprotectant ABC transporter ATP-binding subunit YehX, osmoprotectant ABC-transporter-binding protein YehZ, and osmoprotectant ABC transporter permease protein YehY); betaine biosynthesis from betaine uptake, choline, and glycine (GbcB glycine betaine demethylase subunit B, glycine betaine ABC transport system permease protein, and glycine betaine ABC transport system, ATP-binding protein OpuAA [EC 3.6.3.32]); and synthesis of osmoregulated periplasmic glucans (glucans biosynthesis glucosyltransferase H [EC 2.4.1.-], glucans biosynthesis protein C [EC 2.1.-.-], and glucans biosynthesis protein D precursor) (Supplemental Table S5).

Pathways linked to stress response in termite soils.
Virulence, disease, and defense
Sequence analysis linked many pathways to virulence, disease, and defense in termite mound soil (Figure 6). The predominant ones include cobalt-zinc-cadmium resistance, BlaR1 family regulatory sensor-transducer disambiguation, multidrug-resistant efflux pumps, and copper homeostasis. Other observed pathways were tolerance to colicin E2, bacitracin stress response, resistance to arsenic, fluoroquinolones, cadmium, mercury, chromium, zinc, vancomycin, erythromycin, streptolysin biosynthesis and transport, diphtheria toxin, methicillin resistance in Staphylococci, and bacterial cyanide production and tolerance mechanisms. Genes connected to invasion and intracellular resistance (like internalin A, B, C, D, E, and G), bacteriocins (like ABC transporter ATP-binding protein YvcR, ABC transporter permease protein YvcS, bacitracin export ATP-binding protein BceA, and bacitracin export permease protein BceB), adhesion (like amino-acid ABC transporter ATP-binding protein PebC, 16-kDa heat shock protein A and B, and accessory colonization factor AcfD precursor), and toxins and superantigens (like export ABC transporter ATP-binding protein, export ABC transporter permease, major pilus subunit of type IV secretion complex, VirB2 and streptococcal pyrogenic exotoxin B [SpeB]) were observed in the sequence data set (Supplemental Table S6).
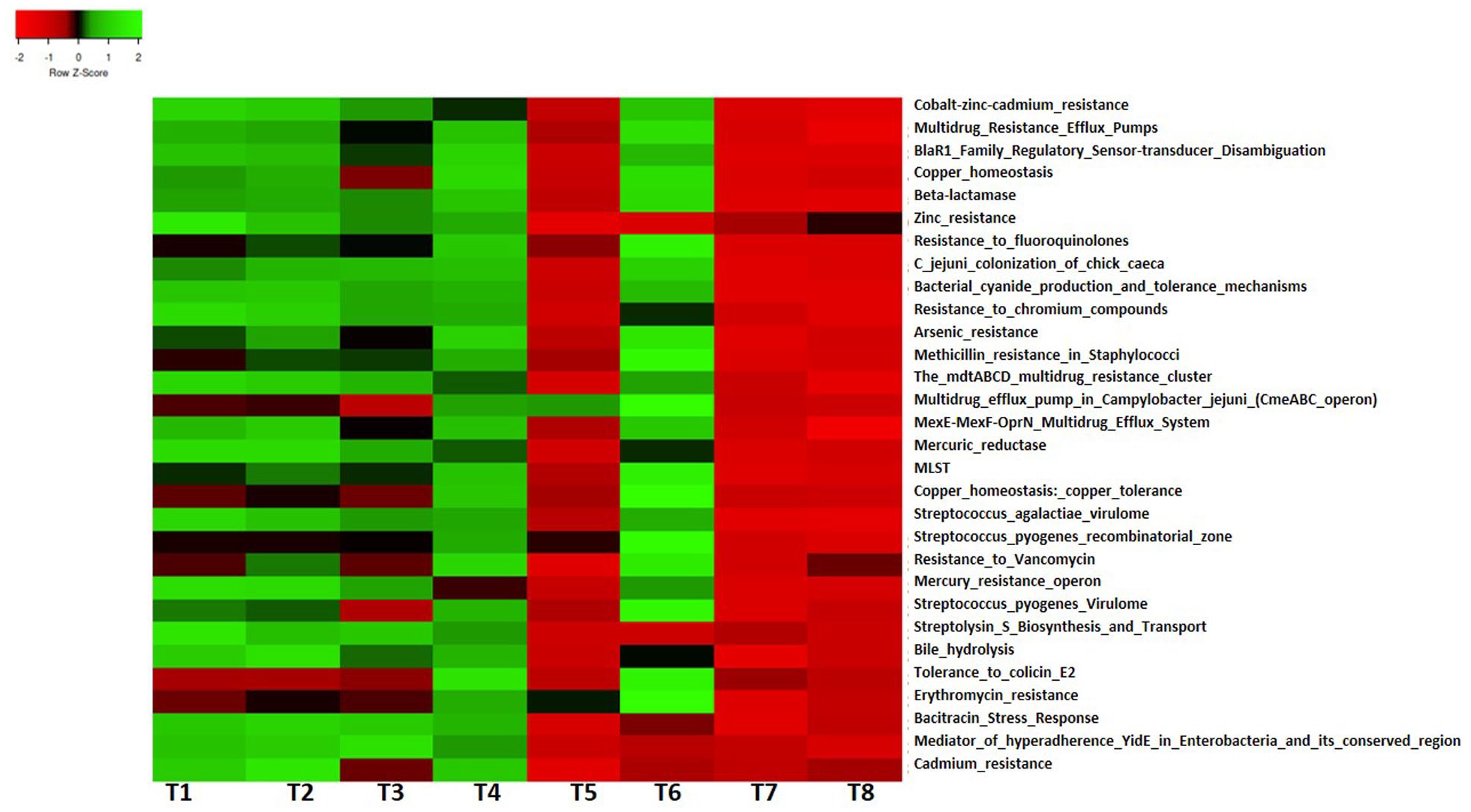
Pathways linked to virulence, disease, and defense in termite mound soils.
Iron acquisition and metabolism
This study identified sequences linked to iron metabolism and acquisition in the termite mound soils (Figure 7). Iron acquisition in Vibrio, siderophore pyoverdine, Campylobacter iron metabolism, heme uptake, hemin, transport of iron, and utilization systems in both gram negatives and positives were the predominant pathways in iron acquisition and metabolism. We also reported the presence of iron-scavenging cluster in Thermus, bacillibactin siderophore, siderophore enterobactin, siderophore yersiniabactin biosynthesis, siderophore pyochelin, vibrioferrin synthesis, siderophore achromobactin, and iron (III) dicitrate transport system in termite mound soils. Gene sequences related to diverse siderophores such as pyoverdine (like pyoverdine sidechain NRPS PvdD, pyoverdine, NRPS modules, and pyoverdine sidechain NRPS PvdJ), bacterioferritin, achromobactin (like achromobactin biosynthesis protein AcsF, achromobactin biosynthesis protein AcsA, and TonB-dependent ferric achromobactin receptor protein), yersiniabactin (like iron acquisition 2,3-dihydroxybenzoate-adenosine monophosphate [AMP] ligase [EC 2.7.7.58, Irp5], iron acquisition yersiniabactin synthesis enzyme [Irp2], and iron acquisition yersiniabactin synthesis enzyme [Irp1, polyketide synthetase]), bacillibactin (like 2,3-dihydroxybenzoate-AMP ligase [EC 2.7.7.58] [bacillibactin] siderophores, bacillibactin synthetase component F [EC 2.7.7.-], and isochorismate synthase [EC 5.4.4.2] [bacillibactin] siderophore), enterobactin (like outer membrane receptor for ferric enterobactin and colicins B, D, enterobactin esterase, and enterobactin synthetase component F, serine-activating enzyme [EC 2.7.7.-]), and pyochelin (like ABC efflux pump, fused inner membrane, and ATPase subunits in pyochelin gene cluster) were found in the data set (Supplemental Table S7).
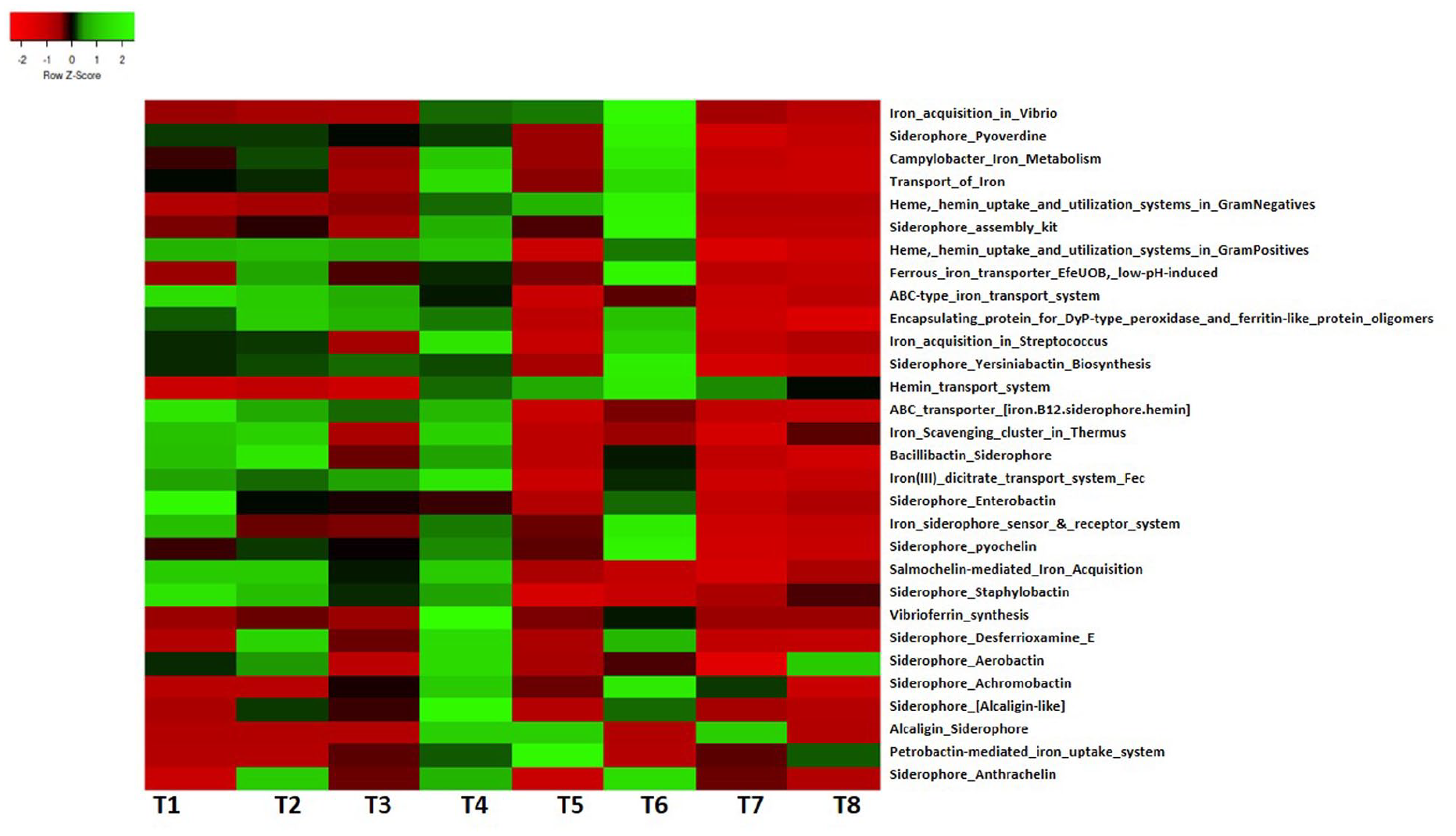
Pathways linked to iron acquisition and metabolism in termite mound soils.
Discussion
Functional characterization of the microbiome in soil from termite mounds is essential for understanding the microbial role in supporting plant development in abiotically stressed, disease-prone, and nutrient-deficient circumstances. 27 This study revealed why plants grown on termite mound soils tend to do well as reported by many local famers.11,12,28 Apart from the fact that termite mound soil contains high soil nutrients as revealed by Ehrle et al 29 and supported by our soil analysis (Supplemental Table S1), it also housed a lot of microbial diversities that are of environmental importance (Figure 1). 7 The dominance of Proteobacteria and Actinobacteria in termite mound soils from this study supported this fact as some strains (Bacillus, Pseudomonas and Streptomyces species) from Proteobacteria and Actinobacteria are recognized to synthesize almost 80% of the total metabolites identified nowadays.30,31 It has also been established that well-known antimicrobial compounds like streptothricin, actinomycin, and streptomycin, which are of agricultural significance, are produced by some species of the Actinobacteria, 32 which was very abundant in termite mound soils from this study.
Identified protein function via KEGG, NOG, COG, and subsystem (Figure 2) revealed community-associated metabolic abilities and useful roles of the termite mound soil microbiome. The communities could function as valued biological sources of novel secondary metabolites with multiple roles like anti-infection, antagonism, antibiotics, and anticancer. Metatranscriptomics research supported the presence of cellular processes, protein families, and particular genes in different environments like soil. 33 Our study revealed a lot of pathways and genes that code for secondary metabolism (Figure 3 and Supplemental Table S3), a unique characteristic of microorganisms to synthesize a variety of metabolites that are significant for self-defense. 32 They also play huge function in microbe–microbe, host–microbe, and microbe–environment collaborations. 34 The presence of genes linked to auxin biosynthesis (a growth regulator influencing the cell elongation and division in plants) in our study shows that termite mound soil housed auxin-producing and auxin-secreting microbiome. Genes that code for cinnamic acid biosynthesis were also identified from our study. This connotes that the microbes in termite mound soil can produce allelochemical phenolic—a chemical that affects metabolic processes, stimulate plant root growth, and promote seed germination. 35 Genes that help in iron acquisition and metabolism were reported from this study (Figure 6). These genes enable microorganisms to utilize iron for stimulating metabolic enzymes/pathways. 36 For instance, high-affinity iron transport systems (siderophores) linked with biosynthetic chelates aid plants, bacteria, archaea, and fungi to withstand iron stress. 37 While microorganisms acquire iron (as siderophores), which is very competitive with the aid of transport systems, plants can also benefit from this acquired iron through microbe–root communication under diverse soil circumstances. 36 Furthermore, our study also revealed the occurrences of several stress-mitigated genes (Figure 4). This means that the microorganisms in soil from the termite mound could have employed them as a survival strategy and environmental performances. For example, glycine betaine (reported from this study) is a key organic osmolyte in bacteria, archaea, and fungi that mitigates diverse environmental stresses such as heavy metals, salinity, high temperature, drought, and ultraviolet radiation. 38 Also, glutathione is a stress-response pathway that militates against oxidative stress and gives fortification against toxic xenobiotics in our environment. 39
The microbial capability to use an aromatic composite by breaking down xenobiotic chemicals is necessary for decontamination of natural environments. 40 The identification of gene sequences linked with pathways for breaking down of chlorobenzoate, chloroaromatic, biphenyl, gentisare, benzoate, quinate, phenylpropanoid compound, naphthalene and anthracene, p-hydroxybenzoate, n-phenylalkanoic acid, toluene, and salicylate ester in termite mound soils (Figure 4) suggests the possibility that the microbial communities in termite mound soil could serve as a useful and promising candidate for biological remediation of soils contaminated by the aromatic compounds.41,42
Our study also witnesses high amount of gene sequences connected to virulence, disease, and defense such as bacteriocins and adhesion (Figure 5). These features allow bacterial communities to shield themselves with ribosomally synthesized antibacterial peptides or bacteriocins. 43 The occurrence of adhesion in soil from termite mounds is indicative of a function in enhancing colonization by bacterial societies residing in termite mounds. The occurrence of gene resistance to mercury, arsenic, chromium, vancomycin, cadmium, cobalt–zinc–cadmium, and zinc in our study suggests the capability of microbes from termite mound soil in bioremediation.44-47
Conclusion
Insight on how diverse microbial populations in termite mound soils impact plant performance and production via metagenomics unlocks innovative prospects for developing ecologically friendly means to maximizing the benefits of microbe-mediated agricultural technologies. Shotgun sequencing of termite mound soils shows high taxonomic richness, with Proteobacteria and Actinobacteria dominating the mound soils. Exploration of our data set revealed the functional abilities of the microbiome connected with the termite mound soils. It also revealed that our metagenome housed a high amount of gene related to secondary metabolism, stress and defense response, iron acquisition, xenobiotic breakdown, normal physiological pathways, and biological remediation. Therefore, the microbiomes are hypothetical to aid development, survival, and growth of the crop in harsh ecological soil conditions. With this knowledge, both innovative culturable approaches should be developed to isolate these microbial strains that can exhibit strong features in hostile circumstances, or we can manipulate organisms with these genes to get new functional benefits.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322231184025 – Supplemental material for Metagenomics Reveals the Microbiome Multifunctionalities of Environmental Importance From Termite Mound Soils
Supplemental material, sj-docx-1-bbi-10.1177_11779322231184025 for Metagenomics Reveals the Microbiome Multifunctionalities of Environmental Importance From Termite Mound Soils by Ben Jesuorsemwen Enagbonma and Olubukola Oluranti Babalola in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
B.J.E. thanks South Africa’s National Research Foundation/The World Academy of Science African Renaissance grant (UID110909). Work in O.O.B.’s lab is based on the support by the National Research Foundation of South Africa (grants ref: UID81192, UID105248, UID95111; OOB).
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the National Research Foundation of South Africa (Grants Ref: UID81192, UID105248, UID95111) granted to OOB.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
B.J.E. managed the literature searches, wrote the first draft of the manuscript, performed the analyses, and interpreted results while O.O.B. conceptualized the project idea, designed the study, enrolled and supervised the B.J.E., thoroughly critiqued the manuscript and proofread and improved the drafts. Both authors approved the article for publication.
Consent for Publication
Not applicable.
Availability of Data and Materials
Sequences used in this study have been deposited in the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) under the bioproject numbers PRJNA526912.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.