
Abstract
Nowadays, the integration of biological data is a major challenge for bioinformatics. Many studies have examined gene expression in the epithelial tissue in the intestines of infants born to term and breastfed, generating a large amount of data. The integration of these data is important to understand the biological processes involved during bacterial colonization of the newborns intestine, particularly through breast milk. This work aims to exploit the bioinformatics approaches, to provide a new representation and interpretation of the interactions between differentially expressed genes in the host intestine induced by the microbiota.
A total of 61 differentially expressed genes (DEGs) in the intestine of newborns extracted from several bibliographic works and databases were annotated for functional analysis using the String software (http://string-db.org/), the Cystoscape software (http://www.cytoscape.org/) and the BiNGO plugin. The latter provided an evaluation of the signaling and metabolic pathways, molecular networks and biological processes for all the used genes.
The analysis revealed that RELA, INS, IRS1, IL1B, and NFKBIA are the central genes in the interaction networks produced. These networks show that the cellular differentiation of the intestinal epithelium and the development of mucosal immunity, are the most affected processes in newborns. Therefore, the global patterns of interactions supports the relationship between breastmilk and the role of the microbiota’s diversity. Ergo all results consolidate the importance of breastmilk and intestinal microbiota in homeostasis.
Introduction
Breastmilk microbiota provides the transient microbiota in the newborn.1-3 This transient microbiota is important for the implantation of the personalized intestinal microbiota of each individual; indeed, it plays a fundamental role in the development of the newborn.4-7 Moreover, breastfeeding has demonstrated its ability to provide a balanced intestinal microbiota to the newborn, thus positively impacting the newborn’s health.8-10 Human milk can stimulate the proliferation of Bifidobacterium and Lactobacillus strains, whose role is to create an acidic environmental rich in short-chain fatty acids (SCFAs) with a protective and nutritive role at the intestinal level.11,12 It has been shown also, that in germ-free rats, Streptococcus thermophilus (transient commensal bacteria) induces the epithelial stem cell differentiation. 13 As well, Bacteroides, very abundant in human colostrum, may have a main role in the early stages of newborn’s gut colonization. 14
Intestinal microbiota is largely studied since 2 decades, and a big amount of data are generated with Omics approaches. However, the interaction between human milk microbiota and newborn’s intestinal microbiota is not completely clear. Other studies are needed to understand the powerful relationship between the human milk microbiota and the stimulation of newborn’s homeostasis. Novel approaches have been developed to study the microbiota using the directed acyclic graphs (DAG) method to facilitate an understanding of the ontological profiles provided by the interaction between the induced genes in the newborn’s epithelium breastfeeding. 15
Ontological studies are becoming essential to understand the complex mechanism placed in the intestine of newborn during breastfeeding. Thus, the biological processes and Top functions can be identified through the predicted networks. The omics technological advances associated to bioinformatics tools allowed to have a global view of the function of genes and their interactions in the cell, in any given context. 16
This study aims to provide a new view of the hierarchical ontology while showing the whole biological processes induced in the host’s epithelium during breastfeeding.
Materials and Methods
This work consists of studying, through a statistical-computing approach, the representation of biological data to make their integration more efficient in the case of DEGs in the newborns’ intestine. For this purpose, we have chosen the scientific publications17,18 which recruited healthy, full-term infants, who were exclusively breastfed at 3 months postpartum.
For that, we have used search terms for the PubMed database (www.ncbi.nlm.nih.gov/pubmed/): Breast Feeding, Gene Expression Profiling, Infant, Newborn, Transcriptome, Feces/cytology, Proteome. Sixty-one DEGs that were significantly higher in term infants, were selected from scientific publications. The databases were consulted (GenBank, www.ncbi.nlm.nih.gov/genbank/; GENE, www.genecards.org/) to assign each gene to its iD and their functional annotation (Tables 1 to 6). The results were then processed by different software packages: the String software (https://string-db.org/), the Cystoscape software (http://www.cytoscape.org/) and the BiNGO plugin.
Differentially expressed genes (DEGs) in the gut of neonates born to term and breastfed, selected from various databases and scientific publications.

Differentially expressed genes (DEGs) in the gut of neonates born to term and breastfed, selected from various databases and scientific publications (continued).

Abbreviation: ARF, alternative reading frame.
Differentially expressed genes (DEGs) in the gut of neonates born to term and breastfed, selected from various databases and scientific publications (continued).
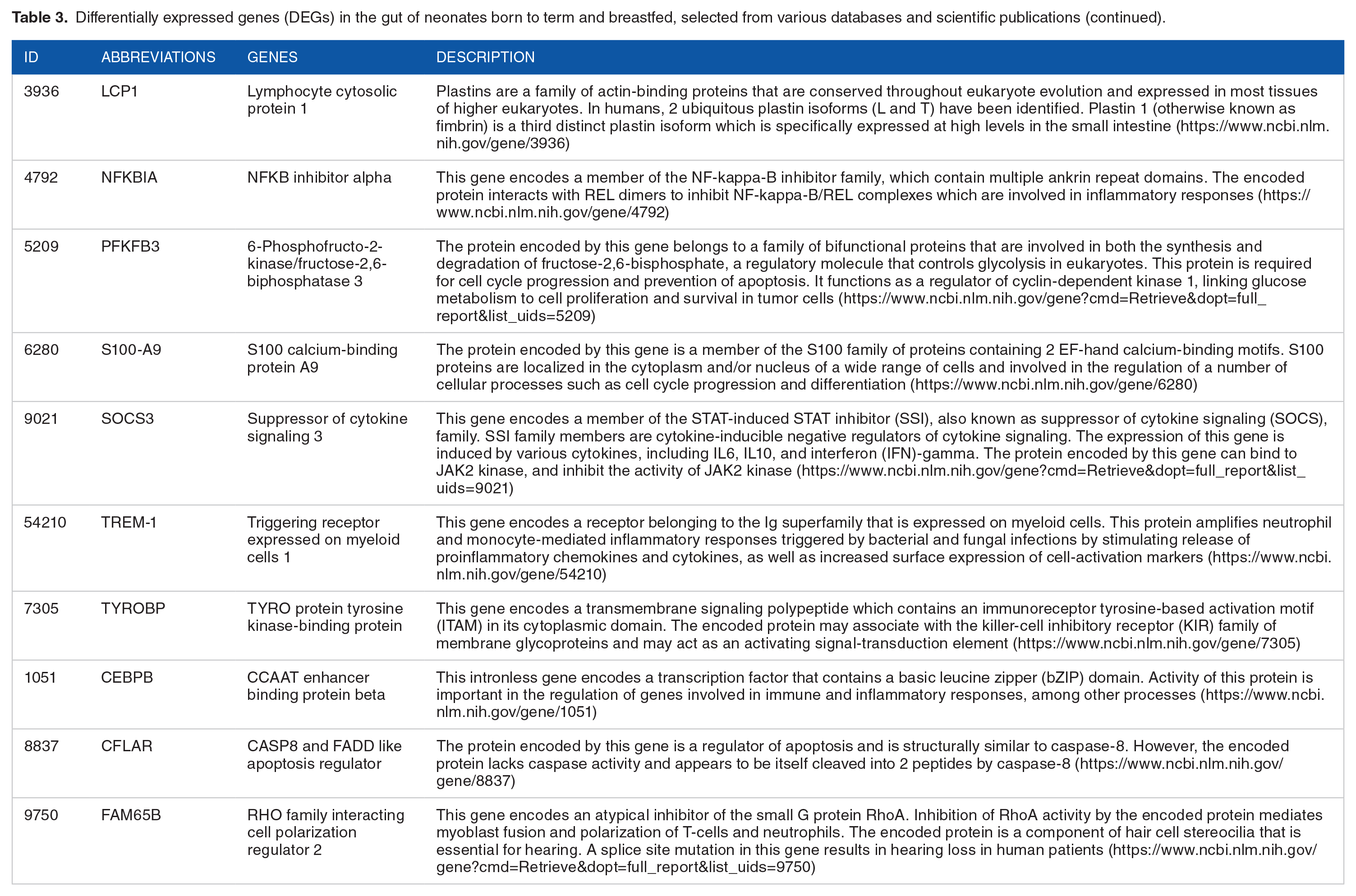
Differentially expressed genes (DEGs) in the gut of neonates born to term and breastfed, selected from various databases and scientific publications (continued).

Differentially expressed genes (DEGs) in the gut of neonates born to term and breastfed, selected from various databases and scientific publications (continued).

Differentially expressed genes (DEGs) in the gut of neonates born to term and breastfed, selected from various databases and scientific publications (continued).

Coexpression analysis by String software
This analysis was performed by 2 functions:
Visualization of interactions between genes by emphasizing particular criterias such as co-occurrence, coexpression, experimental evidence, existing databases, and text mining.
Rich statistical analysis indicates that the terms are classified by their enriched P value. The P value is calculated by a hypergeometric test and then corrected for multiple tests using the Benjamini and Hochberg method.
Ontological analysis by Cytoscape software
The networks generated by string were imported as a pre-existing unformatted array in Cytoscape software.
The network analyzer plugin function provides a network customization. The size and color of the nodes have been customized according to the values of the chosen parameters:—A number of connections of the node with other proteins;—large size and light color for the weak connectivities.
The genes annotated by Cytoscape and selected manually were analyzed by the BiNGO function for network personalization. The network personalization determines a functional profile as DAG interactions. The ontological level “biological process” was chosen as a query for the analysis with the BiNGO plugin function.
Results and Discussion
String results
Sixty-one DEGs (Tables 1 to 6) were imported and analyzed by the String software. Fifty-seven genes were annotated and other 10 genes enriched the networks generated. The results were performed in 2 formats: a network with different confidence indexes (Figure 1) and a network with the different interactions between the proteins (Figure 2). Among the 67 genes annotated on String, 51 have formed a single network and 16 genes remain outside this network. A set of 51 proteins was found to be linked either directly or indirectly through one or more interacting proteins, suggesting the existence of functional links between them (Figures 1 and 2).
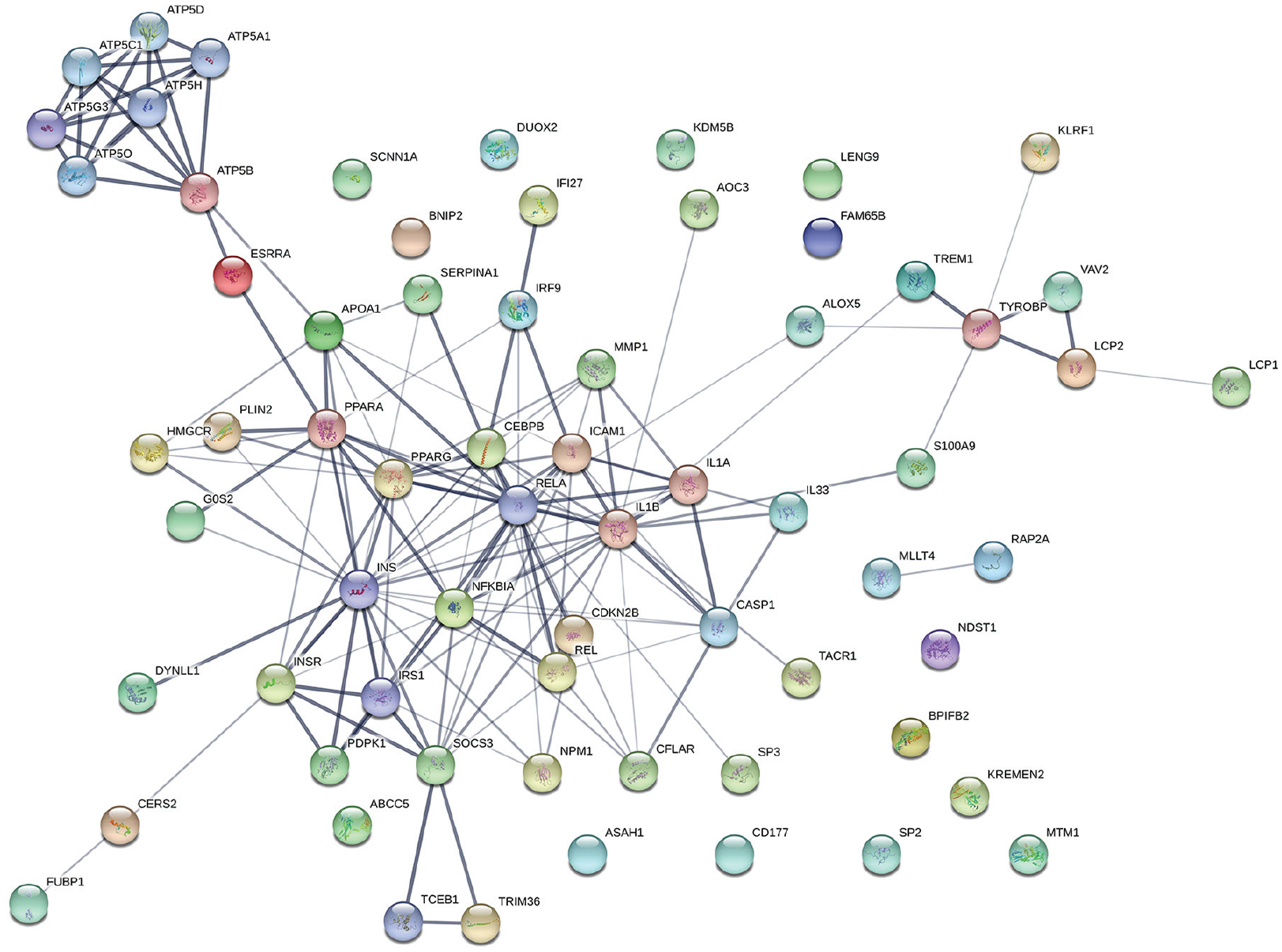
“Confidence view” of the protein-protein interaction network on String.

Types of interactions between the network proteins on String.
The central proteins of this network (Figures 1 and 2) are as follows: IL1β, RELA, INS, IRS1, and NFKBIA.
IL-1 (interleukin-1) production (Figure 1) is mainly regulated by the inflammasome, a multimeric protein complex assembled in response to various inflammatory triggers such as danger signals, microbial toxins, and crystalline substances.20-22 A prototypical complex of inflammasome includes many proteins, among them CASP1 (caspase-1). Cleavage of CASP1 by the inflammasome leads to its activation, which in turn cleaves IL-1β (IL-1 beta).23,24 Interleukin-1β, a proinflammatory cytokine with a wide range of systemic and local effects 25 can modulate the function of both immune and nonimmune cells. Interleukin-1β also promotes T-cell activation and survival 26 and works with other proinflammatory cytokines such as IL-33 (Figure 1) to promote epithelial restoration, repair, and mucosal healing in the intestine. 27 Interleukin-1β can also induce the positive regulation of RELA (transcription factor p65) (Figure 1) and subsequently the activation of the canonical NF-κB pathway, which is necessary for homeostatic regulation of cell death and division in intestinal epithelia, as well as for protection against development of severe acute inflammation of intestines.28-30 Insulin receptor (INSR) mediates the pleiotropic actions of INS (insulin) (Figure 1). Insulin-binding leads to phosphorylation of several intracellular substrates, including IRS1 (insulin receptor substrate 1), subsequently inducing various bioactivities such as growth, differentiation, survival, increased anabolism, and decreased catabolism in many types of cells.31-33
On the contrary, SOCS3 (suppressor of cytokine signaling 3) is regulated by several proteins within the network (Figure 1). It has an impact on multiple signaling pathways, and it is a mediator key of mucosal homeostasis. Suppressor of cytokine signaling 3 is a tumor suppressor, limiting the proliferation of intestinal epithelial cells (angiotensin-converting enzyme [ACE]) in cases of acute inflammation and tumor growth, and plays a role in wound repair.34,35 A group of reactive proteins (black lines) is formed of: ATP5B, ATP5A1, ATP5D, ATP5H, ATP5C1, ATP5G3, and ATP5O, (Figure 1) which have a common role in displaying energy and cells communications. 36 The interconnected proteins within this network (Figure 1) reveal the antiproliferative effect of breastfeeding on the cells of the intestinal epithelium of newborns and the positive effect on cell differentiation. These observations also suggest the involvement of these proteins in metabolism, cell survival, and mucosal homeostasis.
The biological process analysis (Table 7) revealed the presence of different processes, significantly implicated in this network (P value <0.05). The most important processes were:—positive regulation of NF-kappaB transcription factor activity (GO: 0051092);—positive regulation of cellular process (GO: 0048522);—positive regulation of lipid metabolic process (GO: 0045834);—response to organic substance (GO: 0010033).
GO biological processes on String.

Abbreviation: GO, gene ontology.
Cytoscape results
The result network analyzer plugin for network visualization is shown in (Figure 3). The central genes for this network according to the chosen parameters are as follows: INS, IL1B, NFKBIA, and RELA, congruent with the results obtained by the String software.
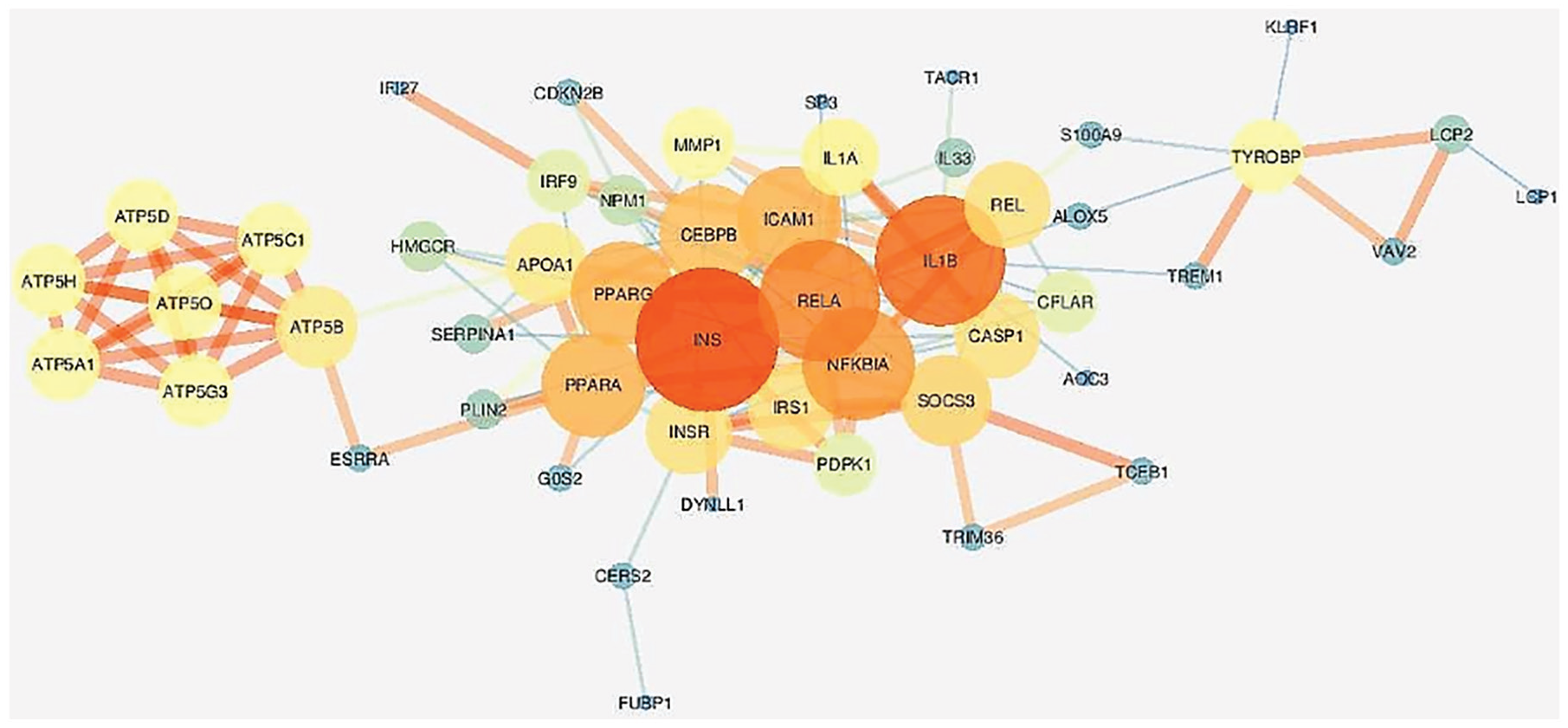
Gene network customized by the network analyzer plugin on Cytoscape.
The GO terms found by BiNGO pluging are displayed as a table of GO terms (Table 8). The functions are grouped into biological processes and the significant ones are as follows: positive regulation of lipid metabolic process, the multiorganism process, positive regulation of the cellular process, and response to an organic substance.
Table of GO terms found by the BiNGO plugin.
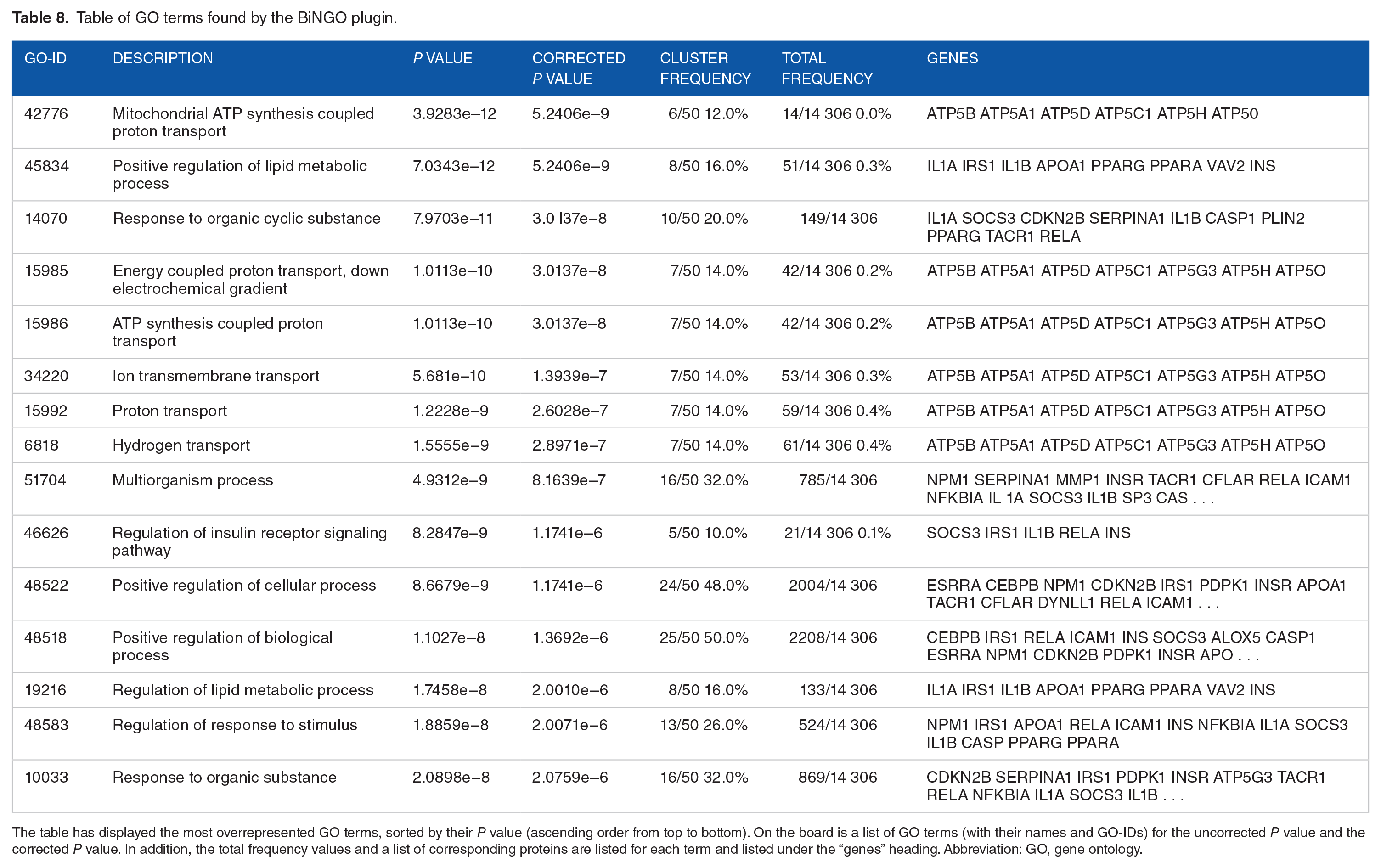
The table has displayed the most overrepresented GO terms, sorted by their P value (ascending order from top to bottom). On the board is a list of GO terms (with their names and GO-IDs) for the uncorrected P value and the corrected P value. In addition, the total frequency values and a list of corresponding proteins are listed for each term and listed under the “genes” heading. Abbreviation: GO, gene ontology.
Positive regulation of lipid metabolic process
Of the 51 genes interacting in the network (Figure 3), 8 genes are annotated in this biological process according to the Table 8. This biological process has a significant P value of 5.2406e–9. The positive regulation of the lipid metabolic process (Figure 4) is related with other term children, the most significant are as follows: the positive regulation of the metabolic process of fatty acids, the positive regulation of the catabolic process of lipids, and the positive regulation of the lipid kinase activity.
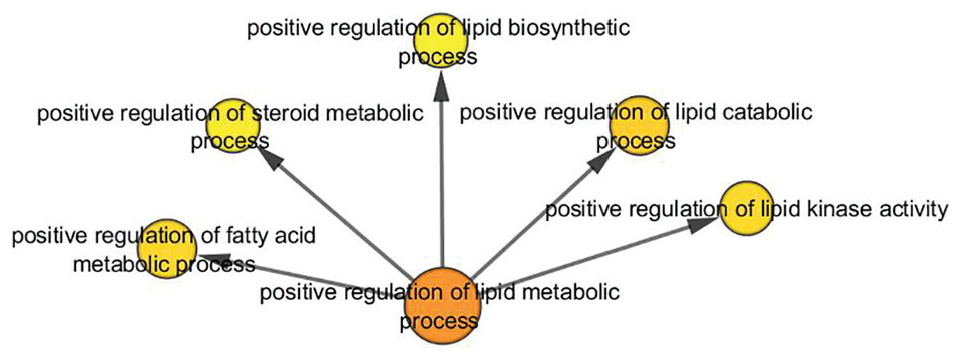
Acyclic-oriented graph of GO terms overrepresented for “Positive regulation of the metabolic process of lipids.” GO indicates gene ontology.
The multiorganism process
Sixteen genes are annotated in this biological process according to Table 8. This biological process has a very significant P value of 4.9312e–9. The multiorganism process (Figure 5) is related with other term children, the most significant are as follows: the response to another organism and the interspecific interaction between organisms.

Acyclic-oriented graph of GO terms overrepresented for “The multiorganism process.” GO indicates gene ontology.
Positive regulation of the cellular process
This biological process has a significant P value of 8.6679e–9, with a ratio of 24 genes annotated (Table 8). The most significant term children from the positive regulation of the cellular process (Figure 6) are as follows: the positive regulation of the cellular communication, the positive regulation of the cellular metabolic process, and positive regulation of organelle organization.

Acyclic-oriented graph of GO terms overrepresented for “Positive regulation of the cellular process.” GO indicates gene ontology.
The response to an organic substance
Sixteen genes are annotated in the response to an organic substance according to the Table 8. This biological process has a significant P value of 2.0898e–8. The most significant term children in this biological process are (Figure 7): The response to the cyclic organic substance, the response to molecules of bacterial origin, and response to the hormonal stimulus.

Acyclic-oriented graph of GO terms overrepresented for “The response to an organic substance.” GO indicates gene ontology.
The BiNGO pluging ontological analysis revealed the involvement of functional network genes in biological processes related to metabolism, communication, and survival of epithelial cells of the gut of newborns. These results are in accordance with the results obtained by String software, showing the positive impact of active foods on the homeostasis of the newborns’ intestines.
Conclusion
The results of coexpression and ontological studies provide insights into global patterns of gene expression in epithelial cells of term infants. The 5 central proteins in the networks (IL1β, RELA, INS, IRS1, and NFKBIA) are the major regulators of 4 significant biological processes. These biological processes induced in the first few months of a newborns’ life have concerned intestinal development, effect of nutrition, and impact of other environmental exposures on the intestinal microbiota colonization. Thus, this study offers a new depiction of the results to allow a better understanding of several interactions and their importance in health homeostasis.
Footnotes
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
F.C., F.B., M.M., Y.S., and B.N. contributed conception and design of the study; B.N. and Y.S. collected data from different databases and scientific publications; B.N. performed the bioinformatics analysis and wrote the first draft of the manuscript; F.C., F.B., M.M., and Y.S. wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Contribution to the Field
Breastfeeding is a strategy favored by evolution, to help our descendants survive and project our genes to succeeding generations. Thus, breast milk is a vector of bacteria in the days and months after birth. Gut microbiota established during these first months of life is vital for infant health and subsequent adults. This work aims to exploit the bioinformatics approaches, to provide a new representation and interpretation of the interactions between differentially expressed genes (DEGs) in the host intestine induced by the microbiota. The results of coexpression and ontological studies provide insights into global patterns of gene expression in epithelial cells of term infants. The significant biological processes induced by the central proteins in the networks have concerned intestinal development, effect of nutrition and impact of other environmental exposures on the intestinal microbiota colonization. So, this study can contribute to a new representation of complex interactions between microorganisms genes and host genome during the development of the intestine allowing a better understanding of several interactions and their importance in health homeostasis.