
Abstract
The exponential growth of genomic data has recently motivated the development of compression algorithms to tackle the storage capacity limitations in bioinformatics centers. Referential compressors could theoretically achieve a much higher compression than their non-referential counterparts; however, the latest tools have not been able to harness such potential yet. To reach such goal, an efficient encoding model to represent the differences between the input and the reference is needed. In this article, we introduce a novel approach for referential compression of FASTQ files. The core of our compression scheme consists of a referential compressor based on the combination of local alignments with binary encoding optimized for long reads. Here we present the algorithms and performance tests developed for our reads compression algorithm, named UdeACompress. Our compressor achieved the best results when compressing long reads and competitive compression ratios for shorter reads when compared to the best programs in the state of the art. As an added value, it also showed reasonable execution times and memory consumption, in comparison with similar tools.
Introduction
Technological advances have led to a considerable reduction of costs in DNA sequencing,1,2 rapidly increasing the amount of genomic data for researchers to process 3 and bringing enormous challenges for its efficient storage and transmission. 4 This trend is expected to continue in the future since sequencing costs are decreasing faster than storage cost. 5
FASTQ 6 and sequence alignment map (SAM) 7 have become de-facto standard file formats in the bioinformatics domain: FASTQ for next generation sequencing (NGS) raw data and SAM for storing alignment/mapping data.8,9 NGS machines produce a file containing typically millions of DNA fragments called reads. 10
A FASTQ file represents reads in plain text, as shown in Figure 1. For every read, the file contains four fields: (1) an identifier with a specific structure that depends on the sequencing platform; (2) a read sequence with the actual DNA bases {A,C,G,T,N}; (3) a separator field (commonly discarded by compressors); and (4) quality scores as ASCII characters in a variable range indicating the probability of a sequencing error in each particular base of the sequence. The range of quality scores also depends on the sequencing platform used to produce the FASTQ file. While for short genome species such as viruses, a FASTQ file can be in the order of tens of megabytes; for humans, it is in the order of tens of gigabytes, containing tens of millions of reads.

Basic structure of the FASTQ format: identifier, read sequence, separator, and the quality scores.
Some of the most important biological sequence databases are doubling or tripling its size annually, 11 a trend that is expected to continue. Currently, remarkable sequencing projects12–15 are set to deliver from terabytes to petabytes of NGS data. From large research centers to smallers bioinformatics labs, efficient storage for the ever increasing data is one of their major IT challenges. 16
Researchers often turn to using compression tools to lower the pressure on storage capacity. Given that FASTQ files are stored as plain text, one can easily rely on traditional general-purpose compression tools. Converting characters into bit streams, 17 using static or dynamic dictionaries,18–20 and performing statistical analyses 21 are strategies implemented by many of these tools that alleviate the problem to some extent. These types of tools have seen widespread use for compressing biological sequences22,23 due to their compatibility, robustness, and ease of use, in spite of certain performance limitations.24,25
In the mean time, domain-specific lossless compressors have been developed during the last decade in an effort to increase efficiency. However, compressing biological sequences is an intensive task which demands significant computational resources. 26 Two different approaches have led this trend 11 : non-referential and referential compressors. Non-referential compressors16,27–35 are commonly easy to use and produce self-contained files, but tend to show modest compression ratios. Referential compressors may demand a more experienced user, but are able to reach higher compression ratios when one choses a highly similar reference.
Despite their potential, referential approaches have not been widely applied to the compression of FASTQ files. One key factor in the design of such algorithms is the need for an efficient encoding scheme that allows representing efficiently the differences between the input reads and the so called reference; to maximize the compression capabilities.
Furthermore, state-of-the-art compressors need to face the fact that sequencing technologies are rapidly advancing to provide ever longer reads. Even though today’s databases contain mostly reads of 100-200 bases of length, the most important manufacturers of sequencing machines have already released models able to produce significantly longer reads, for example, Illumina MiSeq v3 (https://www.illumina.com/systems/sequencing-platforms/miseq/specifications.html), Sanger 3730xl, 36 and Ion Torrent PGM (https://www.thermofisher.com/co/en/home/brands/ion-torrent.html).
In this article, we present a referential compressor for FASTQ files. The core algorithms to achieve compression are based on sequence alignment and an elaborate binary encoding scheme. The main contributions of this manuscript are as follows:
A multi-technique compression scheme for referential compression of FASTQ files. That scheme combines different specific strategies to compress all data streams present in the FASTQ file to guarantee a fully lossless compression.
UdeACompress: a lossless referential compressor for reads. Its core algorithms aim at improving the reads compression ratio for the longer reads of newer sequencing machines.
A comprehensive performance analysis of UdeACompress in both of the aforementioned scenarios/implementations, including compression ratio, runtime, memory usage, and an estimation for a parallel implementation. Furthermore, we explore the effects of the most relevant involved variables in the compression capabilities of our proposal.
Related Work
Here we present the basics of genomic data compression and a brief review of the most relevant tools in the state of the art. While early compressors for DNA focused on genome compression only,37,38 during the last decade, there have been many efforts in developing specialized compressors for different types of DNA data in different file formats. In 2013, the SequenceSqueeze competition focused on promoting specialized compression for FASTQ files due to their relevance. 39 Several lossless non-referential compressors for FASTQ have been released since then16,23,27–35,39–41 and most of them have been reviewed and tested in detail.9,17,25,42 In our previous tests, top performers achieved compression ratios in the range between 4:1 and 8:1, which is still below from what a referential compressor could theoretically achieve. Also, restrictions related to input data features (file size, read size, technology of the sequencing machine), excessive runtime or low compression ratios, have limited the usage and effectiveness of non-referential compressors.
Recently, some non-referential tools for the compression of reads sequences in FASTQ files have been presented with very good results. For the sake of completeness, they will be included in the comparisons of the “Results and discussion” section: ORCOM, 43 HARC, 44 and Assembltrie. 45
Referential compression
DNA sequences from the same species exhibit extremely high levels of similarity. This fact is exploited by referential compression schemes, whose key idea is to encode sequences with respect to another reference sequence(s), achieving very high compression ratios.17,46 Figure 2 shows an example of a basic referential scheme.

Referential compression. A sub-sequence is represented as the pair (x, y), where “x” is the start position on the reference sequence and “y” expresses how many symbols of the sub-sequence are represented. Short differences may be encoded as raw strings.
One of the biggest challenges in referential compression algorithms is to efficiently find long matches between the reference and the read sequence to be compressed. Current approaches use heuristics based on index structures, hash-based structures, graphs, suffix trees, alignment data among others.16,47–50 Once matches and mismatches are determined, another challenge is to find a space-efficient encoding scheme.
A wide range of compression ratios has been reported9,17,51 for reference-based compression. If computational resources and a good reference are available, this approach is ideal for the compression of long sequences or collections of sequences, since very high ratios can be achieved (eg, 400:1). 17 However, it should be noted that decompression requires exactly the same reference used for compression. 23 This is why a referential approach makes sense as long as one reference is used to compress multiple sequences.
Referential compressors for DNA sequences can be divided into different categories. Due to the scope of this article, we will group them according to the characteristics of the data to be compressed.
Genome compression
Two approaches are considered here: the first one is focused on compressing a unique long sequence (a genome)22,24,52–56 and the second approach involves the compression of highly similar collections of genomes.57–62 The level of redundancy in data implies applying different strategies in both cases.
Compression of read sequences along with alignment/mapping information
It refers to compressing file formats that put together reads along with read-to-reference alignment data. This approach commonly takes the input from SAM/BAM files 7 and requires a specific compression approach to target the alignment information. Finally, file formats such as SAM/BAM have a significant amount of additional fields that should be compressed too. Examples of tools that work this was can be found in the references.8,46,63–68
Encoding alignment information is a field of our interest since we selected this approach for the referential compression, but with a FASTQ file as input. We found a very important antecedent in the work of Kozanitis et al
47
in 2011, as they introduced a set of domain-specific referential lossless compression schemes for reads and alignment data that, according to its authors, compressed the read sequences over
Compression of NGS raw data
In this category, we either find (1) multi-file compressors for large datasets of highly related reads or (2) single-file compressors for reads. In both cases, this implies handling short raw redundant sequences, and sometimes the compressor also processes the identifiers and quality scores, although using different approaches. Examples of such compressors include Yanovsky, 69 Daily et al, 70 and Zhu et al. 71
In Kpath, 49 authors combined path encoding, De Bruijn graphs and context-dependent arithmetic coding to offer reference-based compression without the need of a previous alignment. Authors claimed that a high compression could be achieved even if the reference was poorly matched to the reads. Reported results showed that the compression ratio was up to twice better than the best specialized non-referential compressors tested.
Leon 48 proposed the use of a probabilistic De Bruijn graph based on a Bloom filter, representing reads and quality scores as paths mapped in the graph using arithmetic encoding. Reported results showed that the compression ratio of the tool crucially depended on the quality of the reference, which is built from the reads. In overall, that compression ratio was up to 10% higher than the non-referential methods presented in that report.
Even though compressing the three data streams in FASTQ files (namely read sequences, identifiers, and quality scores) is required for a truly lossless compression, few tools offer such capability. The well-known non-referential compressors Quip, 23 Fastqz, 39 and Fqzcomp 39 are able to compress the whole FASTQ and allow to perform the compression in referential mode. However, the achieved compression ratio is not better that their own non-referential counterparts. Recent versions of Leon compress all of the data in a FASTQ file. In 2015, FQZip 72 was presented as a reference-based method to compress the whole FASTQ file, which evolved to a second version of a light-weight mapping model (LWFQZip2 73 ), achieving compression ratios comparable to those of non-referential programs.
In spite of the promising results reported by some FASTQ referential compressors,48,73 we consider there is ample room for improvement given the great potential to achieve higher compression ratios when using a referential approach. Naturally, these expectations are limited by the difficulty of (1) finding the most similarities between the input and the reference, (2) devising an efficient encoding scheme to compress differences that takes into account the trend for longer reads, and (3) integrating in a single tool efficient compressors for the three streams of data in a FASTQ file. The work presented in this article aims at advancing the state of the art of referential compression for FASTQ files considering the aforementioned challenges.
Multi-technique Compression Scheme
We have built a FASTQ compressor that uses multiple techniques to deal with each of the three data streams as shown in Figure 3. Since quality scores play a role in the compression of reads, they are also fed to UdeACompress. After reordering the reads, UdeACompress sends the sorted position of each read to the blocks in charge of compressing the quality scores and the identifiers; to keep the compressed file consistent.

The multi-technique compression scheme: referential compressor for reads sequences, identifiers (IDs) compressor, quality scores (QSs) compressor. Currently, black boxes are implemented using third-party software.
Packing and unpacking
Since the three data streams in a FASTQ file are very different in content, length, alphabet, and the level of similarity among reads; differentiated compression strategies need to be applied. To set things up for the separate processing of data streams, two blocks are placed at the beginning and at the end of the compression process. The unpacking block reads the FASTQ file and creates three data streams for further processing. Conversely, the three compressed data streams are put together in a single file by the packing block.
Reads compression
Read sequences are processed with UdeACompress, the referential compression algorithm we developed, and the core of the multi-technique compression scheme. Details about the development of UdeACompress will be presented in section “Reads compression with UdeACompress.”
Identifiers and quality scores compression
Identifiers have a format that depends on the sequencing platform. They use a wider alphabet and account for less data than the other two streams in the FASTQ file. Usually, they have little variation between reads of the same file, which is is usually exploited using delta encoding approaches. 74
Quality scores are more difficult to compress. They have the same length of the read sequences but use a much larger alphabet that also depends on the configuration of the sequencing platform. There is currently research concerned specifically on quality scores compression.75,76
Since this article is focused on developing a referential compressor for the read sequences stream, the compression of identifiers and quality scores is performed using third-party software. Considering the compression ratio, running times, and software dependencies as reported in a previous study, 25 we selected QUIP 1.1.8. It compresses consecutive identifiers using delta encoding and quality scores with Markov chains. 23
Reads Compression With UdeACompress
UdeACompress performs a referential compression of the read sequences as the core of the multi-techique compression scheme. Our approach is based on the hypothesis that encoding the differences in the alignment between each read and the reference is a powerful strategy for referential compression. The algorithms are aimed at increasing (1) the quality of the alignment according to our specific compression goals and (2) the encoding efficiency.
UdeACompress first performs a specialized alignment between the input reads and the reference and then sorts the reads according to their mapping position. These positions are encoded into a binary map and the alignment data are binary encoded. Finally, as some reads do not align to the reference, they are compressed separately using a low-level compressor. The inner structure of this module is presented in Figure 4.

UdeACompress block diagram. (1) Specialized read-to-reference alignment, (2) reads sorting, (3) reads encoding, and (4) low-level compression for encoded data and unmapped reads. The sorted positions resulting from the sorting step are used for the compression of identifiers and quality scores as well, to guarantee a correct decompression. Currently, black boxes are implemented using third-party software.
Read-to-reference alignment
Sequence alignment is a very common procedure in bioinformatics. It aims at finding an approximate matching to maximize a similarity score based on some criteria that varies according to the goal. In UdeACompress, every read is aligned against the reference genome to find the region that is most similar to the read sequence, which allows for an efficient compression.
The aligner is based on the seed-and-extend strategy, 77 which provides noticeable performance and accurate results. Multiple substrings are extracted from the read as potential seeds for the alignment. After an exact match is found through an FM-index78,79 that privileges bases with high-quality scores, an alignment is extended in both left and right directions using a modified Needleman-Wunsch algorithm. 80
The aligner outputs a set of instructions describing the transformations needed to obtain the original read from a specific position in the reference. Those transformations (named mutations) are represented with several alphanumeric fields in SAM notation style, commonly used to express alignments. In such abstract notation, data are distributed among several fields which spans over a wide alphabet, containing positions for every mapping, the direction of the matchings and details about each of the mutations to be performed: offsets (displacement between changes), the type of operation that must be executed, and the corresponding target base. In certain cases, those fields must be analyzed together to get unambiguous and precise information.
The few reads that cannot be aligned to the reference are passed as they are, and we call them unmapped reads. Specific details on the design, implementation, and optimization of the aligner can be found in Lotero et al. 81
Reads sorting
Sorting reads is a strategy that many programs have applied to increase effectiveness in sequence compression,9,41,43–45 by putting together reads that are more similar. Although the original order is lost, other works have already discussed how this does not affect most post-processing tasks of the uncompressed FASTQ file, given that their originally placement is anyway arbitrary. 9 After the alignment, UdeACompress sorts the reads according to their mapping positions, as a pre-requisite to build the map required for encoding.
For the sorting we implemented and tested different algorithms, which led us to use a least significant digit radixsort, since it performed significantly faster than other strategies. UdeACompress sorts the indexes that represent each read, which is used not only for encoding the alignment instructions, but for compressing the identifiers and quality scores as well.
The sorting algorithm performs three main steps iteratively: building a histogram, exclusive prefixing, and placing indexes into a sorting location. The histogram and the placing steps depend on the amount of reads in the input (n), and the process is repeated according to the maximum number of digits of the biggest index (k), which leads us to a complexity of
After this process, mapped and unmapped reads are separated, the alignment instructions are binary encoded, and the unmapped reads are compressed with a low-level compressor.
Encoding
The encoder in UdeACompress produces a space efficient binary coding of the alignments and was inspired by the work of Kozanitis et al. 47 Two data structures form the code: a binary map of the alignment positions and the instructions array. This approach is conceived for files with fixed length reads, which is a trend in sequencing machines. The code is also designed to be further compressed in the so called low-level compressor block.
The code format is shown in Figure 5 and the algorithm to produce it in 1. The next sections explain the details.

Instruction encoding. (I) A single map with as many bits (n) as bases in the reference to indicate the matching positions. (II) A three-field instruction for each read: (a) a mandatory 4 bits PRELUDE for describing the matching types, (b) a 10 bits OFFSET to position each mutation, and (c) the description of the MUTATION itself (6 bits). The OFFSET and MUTATION fields are not required for exact matching types.
Instruction encoder.

Map builder
The map, shown in Figure 5 with an example reference above, is a binary array with as many bits as the reference. It only has 1’s in the positions where one or more reads map (the start of an alignment). This map definition aims to reduce the cost of representing the mapping positions of the reads in the reference and is the reason why the reads must be previously sorted. No matter how many reads there are in the input, the map size only depends on the reference length and is shared by all reads.
Alignment instructions encoding
It is focused on generating a succinct representation of the alignment instructions in a binary space, while also producing a uniform distribution of bits to benefit more from the low-level compression.
The first of three fields is a mandatory and fixed sized PRELUDE, as shown in Figure 5. It uses 4 bits for storing the matching information per read and it is the minimal representation for a read matching in this model. The first bit of the PRELUDE (called MoreR) indicates whether the next read maps to this position as well, or not. The next 3 bits encode eight different kinds of matchings according to Table 1 (Match). The basic matchings (forward and reverse) could be exact or approximate (with at least one mutation), for a total of four cases. Since exact matchings are the cheapest to store, our strategy to increase its probability of occurrence was using an extra bit to incorporate two additional types of matchings well known in bioinformatics, but not commonly used in alignment: complement and reverse complement. This will also help increasing the amount of mapped reads, which are compressed more efficiently than the unmapped ones.
Matching codes.

In complement matchings, each of the bases in the reference sequence must be substituted by its biological complementary base (see Table 2), with N’s not having a complement.
Bases complement.

The prelude is enough for storing the exact matches, but efficiently storing the mismatches in the approximate matchings is the tricky part. The other two fields in the instruction coding are used for that purpose: OFFSET and MUTATION. These two fields only appear in the coding of reads that present mutations.
The 10 bits OFFSET represents the shift between the last mutation (or the beginning of the read sequence if there is no previous mutation) and the place where the current mutation starts. The 10 bits reserved for the offset guarantees the capability of UdeACompress to support reads of up to 1024 bases, given the current trends in sequencing technology. 82 In consequence, this field has the highest storage cost.
The third field of the instruction is called MUTATION as it describes in detail the type of transformation required to obtain the read. We use the first bit of this field to indicate whether this is the last MUTATION of the read (LastM). The next three bits describe the operation (Oper) to be applied. We defined eight different types of operations (see Table 3) based on the mismatches and gaps commonly used in bioinformatics: substitutions, deletions, and insertions. Finally, we use the last two bits to express the base required to perform the operation (Base). Since we only had two bits to express five possible base values (A, C, G, T, N), we store the distance (to be precise, the distance—1) between the base in the reference and the target base in the read, according to the scheme in Figure 6.
Probability of mutations occurrence.


Circular base distances scheme.
Bases are needed only for single insertions and for substitutions, but in some cases of insertions the target base may not correspond to a base in the reference (eg, in insertions at the beginning or at the end of the reference), hence representing base distances is not possible. To overcome this issue, we separated the regular bases insertions (with target bases: A, C, G, T) from the case of N insertions (which are more common) as different operations. In this scheme, for regular bases (from now on, insertions), the number in the base field represents directly the letter of the target base to be inserted, and for insertions of N (from now on, N insertions), both bits can be omitted or be set to zero.
Clearly, the more mutations per read, the lower the compression ratio. Therefore, we applied the following strategies:
Selecting the least possible number of mutations in the alignment: We influenced the aligner so that an exact matching is always selected if possible. If there are several different matchings, the one with least mutations is selected. Additional matchings previously introduced aim to achieve this goal as well.
Reducing the number of instructions required to express consecutive mutations: After statistically analyzing the most common operations in the alignments of our dataset, we defined a set of additional operations to describe contiguous mutations through a single operation (see Table 3). We complemented the four operations already considered (substitutions, deletion, insertion, and N insertion) with four proposed contiguous operations based on the two most common biological mutations (substitutions and deletions). The four “contiguous mutations” most likely to happen were: Double, Triple, and Quadruple Contiguous deletions, and the Contiguous Repeated Substitutions (substitutions in consecutive positions with same target base). In addition, based on those probabilities (Table 3), we assigned the most efficient binary representation to the most common operations.
Preferring operations that required fewer bits: We defined categories of operations according to the gain in storage saving, to skew our specialized alignment and get optimal results (see Table 4). If there are several alignments with the same amount of mutations for a read, the one using less bits is chosen.
Penalty categories.

An example of instruction encoding
In Figure 7, we present an example of representing three reads using our encoding model.
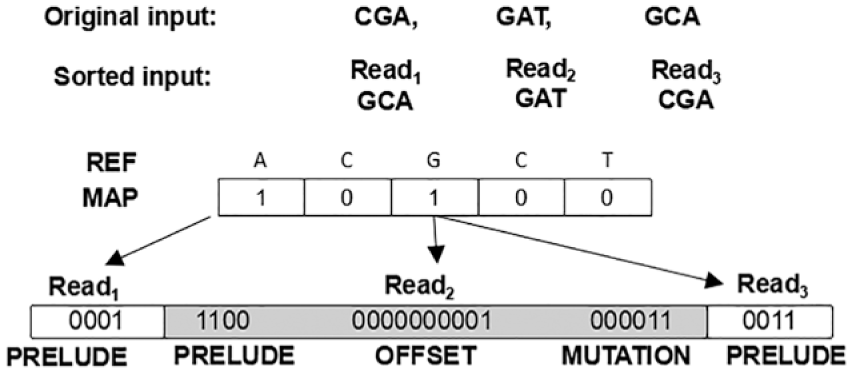
Example of instruction encoding to represent the three reads shown.
At the top of the figure, we observe the original input reads, and below we see them sorted according to the mapping position. The map has 1’s in positions one and three because those are the only locations where reads map. The first mapping position corresponds to
Implementation issues
UdeACompress was implemented in ANSI C. The map, which could require from tens of thousands to hundreds of millions of bits, is stored as an array of 64 bits integers to minimize memory access. For the instructions, we use instead an array of 8 bit integers to provide a finer granularity that facilitates future parallel versions of the algorithm. For this reason, we use the 8 bits of the OFFSET field to store the 8 least significant bits (the offset suffix) of the whole offset. The two most significant bits from the third field are used to store the two most significant bits of the OFFSET (the offset prefix). This division lies in the fact that most of the times the bits of the offset prefix will be zero so there is no need to use them. This also brings a more uniform bit distribution.
The exact cost of representing an instruction will depend on the implementation approach and the hardware restrictions. Common hardware forces fixed size definition for data types, but specific hardware could allow particular sizes for user-defined types, with a great impact in the cost of storage of an instruction.
If the encoder is implemented in a fixed data type size environment, grouping together all the preludes in the same array allows for storing two preludes in a single byte, and for every OFFSET-MUTATION pair 16 bits are needed. The implemented strategies were oriented to require the minimal possible of bits to store each mutation, but in this current implementation saving less than 8 bits in the encoding does not result in a direct reduction of the storage space. However, the resulting padding zeros will benefit the low-level compression.
Low-level compression
The low-level compression block is meant to compress the unmapped reads and to further compress the encoded instructions of the rest of the reads. For the unmapped reads, we chose to use bzip2, the best performing tool for the task according to a previous thorough study. 25 For the compression of the binary instructions, we tested many options but concluded again that bzip2 provides the best balance between speed and compression capabilities.
Bzip2 83 is based on the Burrows-Wheeler transform (BWT) 84 combined with Huffman coding compression. 21 This means that the symbols within the sequence are permuted to increase the repetition of certain sub-chains, which are represented more efficiently according to their probability of occurrence. The BWT transformation improves the compression with a simple reversible method, which is convenient for problems with reduced alphabets but huge amounts of data. We used the low-level interface of the library libbzip2 which is the current API of the bzip2 compressor.
Results and Discussion
In this section, we present the results of multiple performance tests applied to the implementation of UdeACompress and its integration with other modules of the multi-technique compression scheme. We used six different datasets to compare our algorithms in terms of compression ratio and speed against the best specialized compressors in the state of the art.
Datasets and tested programs
We selected six FASTQ files: three plants and three bacteria; details about each dataset can be found in Table 5. This dataset was chosen to have a variety of species, amount of reads, reads length (Illumina style), and reference file size. We have used the formal reference of each FASTQ as provided by the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra). In some of the files, the ID field was originally replicated in the comment field of each read. It was removed to achieve more accurate compression ratio results.
Dataset description.

In addition, an extensive set of additional tests was performed using simulated data, whose characteristics are described in detail in section “Simulated datasets tests”.
We reviewed tens of algorithms and tools for FASTQ compression from the state of the art, but decided to include in the performance study presented in this article, only those that showed the best results. In Table 6, we summarize relevant information about each of the selected programs: the approach used for the compression, the target data, and the number of threads used by default. All of the programs were configured in lossless compression mode and the remaining configuration parameters were left at their default values. Even when Quip and FASTQZ allowed enabling a referential compression mode, in our previous tests, it was evident that such option did not improve significantly the compression ratio while increasing the execution time considerably; for that reason, we discarded such configuration.
Compression software description.

All the tested programs had default multi-threaded execution with different levels of parallelism as shown in Table 6. When necessary, we configured the evaluated applications to force them not to keep the original order in the input reads.
The only program in the literature that is directly comparable to UdeACompress is LWFQZIP2, since it performs an alignment-based referential compression of FASTQ. It is therefore the only possible fair comparison of quantitative results. We present comparisons with the other approaches, as mere reference points and to provide a wider perspective for the reader. In addition, we must make clear that ORCOM is not directly comparable to any of the tested programs since it is a reference free compressor designed for large collections of FASTQ files, instead of compressing a single FASTQ file. However, given its relevance in the state of the art and its impressive performance, we decided to include it here. In the case of Kpath, which generates a file to preserve the reads original order, this size was excluded of the reported compression ratio. We experienced some problems executing Assembltrie, which compressed only two of the six files in our dataset.
Tests were performed on a server with two Intel(R) Xeon(R) CPU E5-2620, 2.10 GHz, 15 360 K cache, for a total of 12 cores and 24 threads, 40 GB of RAM in a shared memory architecture, a 1 TB SATA disk at 7200 RPM, and using Centos 7 OS (64 bits). We report the minimum of the execution time of three replicas performed.
Parallelism
Since the current implementation of UdeACompress runs in single thread mode, we decided to develop a parallel model that allowed us to estimate more comparable speed metrics with respect to the other tools that all support multithread execution. In such a model, we only considered the straightforward parallelization of the most compute intensive tasks that are known to be parallelizable.
Figure 8 shows a simplified block diagram of UdeACompress with profiling information. The profiling data were obtained as an average of testing all the FASTQ files in our dataset. We used gettimeofday() which has microsecond precision, observing no significant variation among the replica tests. Since there are only memory and disk operations in the packing/unpacking steps, we did not not include such functions in this profiling. The decompression task was not considered because the structure of our array of encoded instructions demands that decompression has to be performed sequentially.

Profiling of the sequential version of UdeACompress. Boxes show the percentage of time consumed by each function. Black boxes correspond to third-party software. IDs Comp indicates identifiers compression; LLC EMR, low-level compression of the encoded mapped reads; LLC UR, low-level compression of unmapped reads; QSs Comp, quality scores compression.
There were three blocks consuming 80% of the total time of UdeACompress: (1) the seed calculation performed during the alignment, (2) the compression of quality scores, and (3) the encoding of the instructions. Since quality scores compression was performed by a third-party software, we did not consider it as an immediate choice for parallelization. Equation (1) presents an analytical model to estimate the speedups resulting from parallelizing the seed and encode processes. In the following figures of performance, the blue dashed line bar (named UdeACompressP) right next to the UdeACompress blue bar represents this estimation

Compression ratio
In Figure 9, we show the compression ratio, that is, the ratio between the FASTQ file size and the compressed file size, achieved for the six FASTQ files in our dataset.

Compression ratio for FASTQ files.
Results show UdeACompress achieves similar or better compression ratios than the best state of the art programs, with an improvement between 4% and 27% respect to the second best program for three of the input file tests. In the rest of the datasets, the maximum difference between UdeACompress and the highest compression ratio is only 14%. In five of the six datasets, we achieved higher compression ratios than the rest of referential FASTQ compressors, and in the case of the exception, the best compressor is only 2% above.
Figure 10 shows the compression ratio corresponding to the read sequences only, without taking into account the identifiers and quality scores. This experiment allows us to evaluate in more detail the capabilities of the algorithm developed, given that identifiers and quality scores are compressed using third-party software.

Compression ratio for the read sequences. The bar filled with inclined lines (UdeACompress MR) represents the compression ratio of UdeACompress when discarding the unmapped reads.
In such tests, UdeACompress achieved a high compression ratio for two of the largest inputs. In the other cases, its compression ratio was lower. Our compressor gets closer to the average compression ratio with bigger inputs. Exploring the files content, we found that the main reason for the good performance of UdeACompress for the third input is that it contains a higher amount of consecutive N’s, which is effectively harnessed by our encoding scheme.
The inclined lines bar in Figure 10 shows the compression ratio of UdeACompress when processing mapped reads only, which naturally reflects a much better performance of our method in such scenario. The negative impact in our design becomes evident when comparing both UdeACompress bars. Our compressor is significantly affected by the compression of unmapped reads, which is done using a general-purpose compressor (bzip2). It decreased the compression ratio (in average) up to 50%, respect to the compression of mapped reads only. The other programs in this experiments use methods that are not affected by the phenomenon of the so called unmapped reads. The efficient compression of unmapped reads requires a very different approach that is to be included in future versions of UdeACompress.
We can analytically determine some of the factors that may affect UdeACompress performance:
The amount of unmapped reads,
Read lengths, since at least 2 bits in the offset are permanently underused in each mutation when reads are short,
The amount of mutations per read,
The fixed sizes for the software data types which limits the benefits of bit encodings that do not match the established sizes and,
The referential method used, as it is, does not fully harness possible inter-reads redundancies since it focuses on read-to-reference encoding.
It must be noted that the performance of UdeACompress was always above the performance of all referential compressors for FASTQ files.
Throughput
It is expected that high compression ratios and fast performance are conflicting goals. Even though we were focused on compression ratio, we also wanted to compare the execution time considering that it is a very important usability factor. We present the following results in terms of throughput, defined as the amount of megabytes per second (MB/s) processed for each program during the compression and decompression.
Figure 11 shows throughput during compression of the FASTQ files. Although the sequential version is outperformed by most of the others, the parallel estimation tell us UdeACompress throughput would be similar to the fastest programs available. Our algorithm is sensitive not only to the size of the input file (length and number of reads) but also to the size of the reference, increasing significantly the amount of memory and CPU required. On average, our sequential and parallel algorithms processes data at around 3-4 and 10-20 MB/s, respectively.

Throughput during compression of FASTQ files. The dashed line bar (UdeACompressP) represents the estimated throughput of a parallel implementation of UdeACompress using the 24 threads available in our setup.
Figure 12 shows the results of compressing the read sequences only. UdeACompress was faster than Kpath but slower than the other applications tested, at a variable rate. This is explained mainly by the fact that as the input and the references increase their size, the alignment process takes longer and UdeACompress speed decreases significantly. Furthermore, the main goal of this version of UdeACompress was to achieve high compression ratios.

Throughput during compression of the read sequences. The dashed line (UdeACompressP) represents the estimated throughput of a parallel implementation of UdeACompress using the 24 threads available in our setup.
In the two compression scenarios presented, a parallel version put us in a competitive position with the fastest programs available. Limitations in the performance improvement of UdeACompressP in Figure 12 show the impact of the low-level compression which is also performed sequentially since there is no parallel version for the bzip2 API yet.
Decompression results corresponding to all of the streams inside the FASTQ file are presented in Figure 13. UdeACompress shows an average throughput of 11.5 MB/s being faster than the other alignment-based compressor (LWFQZIP2).

Throughput during decompression of FASTQ files.
Throughput during decompression of the read sequences only is in Figure 14. UdeACompress behaves consistently, with no big variation at an approximate rate of around 11 MB/s, in overall

Throughput during decompression of the read sequences.
In Table 7, we summarize the measurements related to the results presented in this section.
Summary of performance results.

Abbreviations: CR, compression ratio; CT, compression throughput; DT, decompression throughput; T, max number of used threads; UdeaC, UdeACompress; UdeACMr, UdeACompress performance on mapped reads only; UdeACP, UdeACompressP (estimation of performances of a parallel version).
Bold numbers represent the respective maximum.
Peak memory usage
Although not part of our main goals, we present here experiments to measure the memory consumption of UdeACompress and the other applications for the sake of completeness. Table 8 presents the peak memory usage as part of the analysis commonly carried out in this field.
Peak memory consumption during compression and decompression (MB).

Below each dataset identifier, we show the total size of the full FASTQ input file and the size of the read sequences only.
To measure the peak memory consumption (in MB), all programs were executed using their default configuration, both during compression and decompression. In terms of memory usage, UdeACompress is not as thrifty as the other compressors for the whole FASTQ file. However, in the field of read sequences compression, it is common to expect much higher memory requirements, due to the complexity of the techniques and data structures involved. Both during compression and decompression, UdeACompress could be classified among the most memory demanding tools, along with Kpath and Assembltrie.
The amount of memory required by UdeACompress during compression is almost proportional to the input of the FASTQ file. In overall, during decompression, UdeACompress demanded around 25% less memory. One of the issues to be tackled in future versions of UdeACompress is finding efficient ways of handling the data structures related to the alignment and the reads encoding, where the most memory is consumed. Particularly, the biggest data structures are required during the seeding and extend phase of the alignment, and in the process of encoding the alignment data to generate the binary instructions. Despite exhibiting high memory usage compared to other tools, the absolute values measured are within the memory capacity of high-performance machines commonly available in bioinformatics research centers.
Simulated datasets tests
Since UdeACompress encoding scheme is designed to be more efficient when handling longer reads than what is commonly found today in public databases (a few hundred bases), we decided to analyze its compression potential over inputs with longer reads and varying the amount of mutations per read. Since these experiments are only related to changes in the read sequences properties, we only report the compression ratio of the read sequences, and we only compare with the applications compressing such stream only.
Subsequently, a set of simulated data files was created to test the performance of UdeACompress under different scenarios. The goal of these experiments was to test compression capabilities of UdeACompress measuring: (1) the effect of the read lengths variation, since this was the main factor considered in our instruction design; and (2) the effect of the number of mutations per read, as the instruction sizes grow when the number of mutations increases. For such dataset, we built files with up to 6 GB of read sequences only, generated from a human reference to expand the range of species included in our evaluation.
Experimental setup
We built a tool using ANSI C to create simulated FASTQ files in Illumina style, considering a set of relevant parameters: read length and maximum number of mutations per read (see Table 9). Also, we defined probability functions to calculate the matchings, mutations, bases, offsets and to calculate the maximum number of mutations per read. Outputs include the required identifiers and quality scores, but we used empirical values for the generation of such fields since they are meant to be discarded by all the compressors in these tests. To make a fairer comparison, only classical matchings (forward and reverse) were used to generate the simulated data files and not skew the results to our benefit. All the simulated datasets were generated using a human reference, the Homo sapiens chromosome 6, GRCh38.p12, primary assembly (https://www.ncbi.nlm.nih.gov/nuccore/NC_000006.12?report=fasta), downloaded on June 14, 2018.
Simulated tests configuration.

Mutations were adjusted to an exponential distribution. 47 After a previous statistical analysis of FASTQ files, we found that a typical upper bound for the quantity of mutations per read is below 10% of the read size, so we generated a maximum number of mutations between 0% and approximately 10% of the read length in order to test the performance of UdeACompress in situations of non-optimal alignment between the reads and the reference. Probabilities of occurrences for bases and mutations were established according to the values presented in Table 3.
By default, the maximum of number of mutations per read was set to 10% of the read length; read length was set by default to 1024 bases, and the calculated coverage of the simulated data was approximately 70×, according to recommendations (https://www.illumina.com/science/education/sequencing-coverage.html). 85 Unmapped reads were not considered in these tests since their processing corresponds to a third-party software.
Although we intended to test the same compressors for read sequences presented in section “Compression ratio”, our tests revealed that ORCOM was not able to compress any of the files with large reads. Also, the documentation of HARC and Assembltrie states they cannot handle large reads either. Only Kpath matched the requirements to compress the simulated data, which highlights the contribution of our approach.
Read lengths effect
The first factor to impact UdeACompress performance is the read length. Its effect on the compression ratio was measured while varying the maximum number of mutations per read, as shown in Figure 15. The maximum percentage of mutations was restricted considering only 10% and 25% mutations per read. Results are presented in Figure 15.
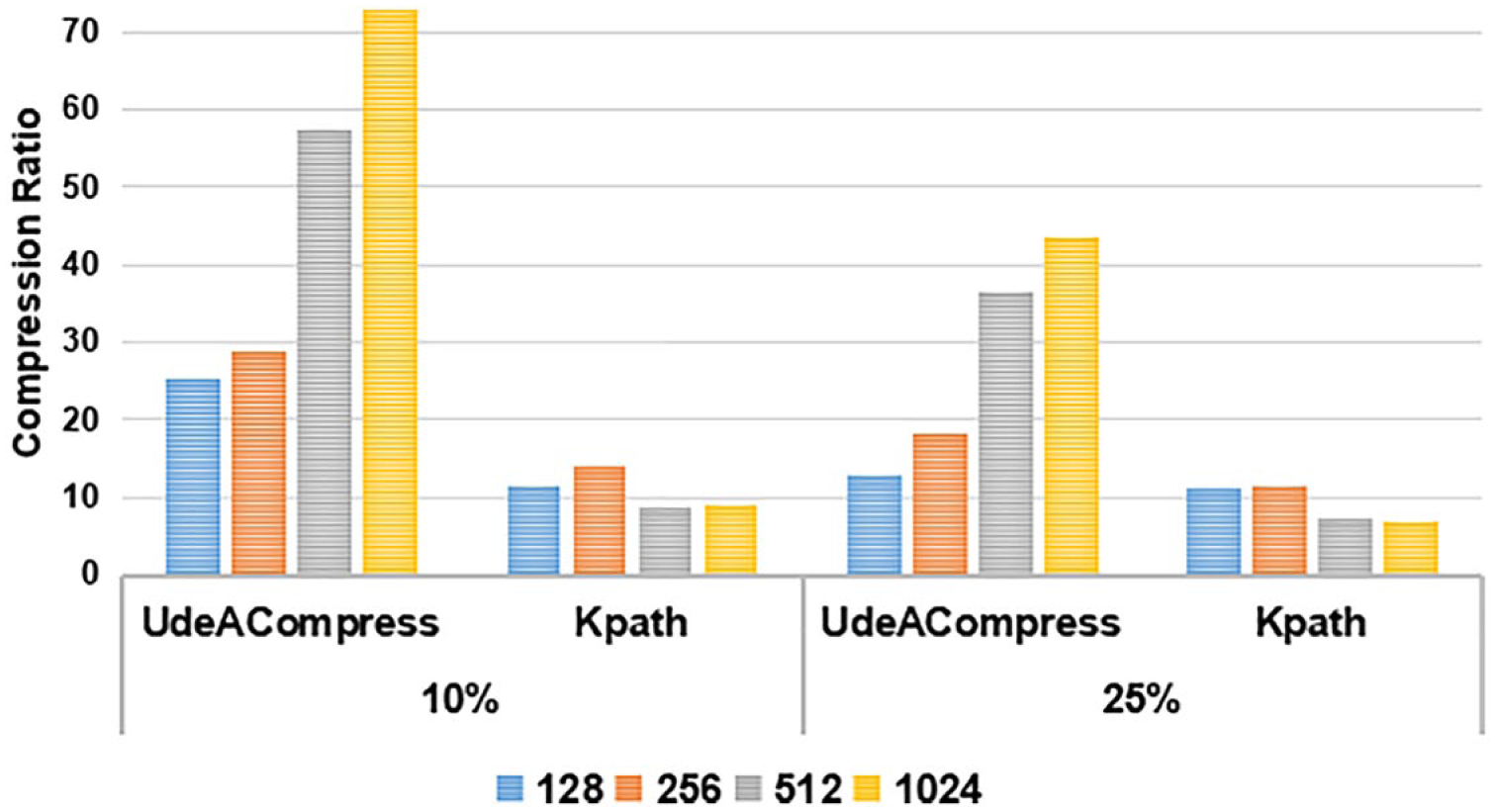
Compression ratio of the read sequences through the variation of the read length and the percentage of mutations per read.

Compression ratio of the read sequences through the variation of the maximum percentage of mutations per read.
The compression capability of UdeACompress significantly increases as the reads get longer and the percentage of mutations decreases. This is expected since with longer reads, our instructions encode more information using the same amount of bits, and the impact of storing the map is reduced because the same structure is used to represent more data.
Figure 15 also shows a scenario where shorter reads (128, 256) have a very high percentage of mutations (25%), which impacts negatively our referential compression. Even in such cases, the performance UdeACompress is still acceptable compared to the other evaluated application. Considering a significantly high maximum percentage of mutations (as 25%) and reads of length equal or superior to 512, UdeACompress achieves a compression ratio between
Effect of the maximum number of mutations
For this test, we generated reads with a maximum amount of mutations between 0% and 25%. It must be noted that because of the exponential distribution, the percentage of reads with exact matchings (zero mutations) is always higher than any other number of mutations probability. Also, since contiguous mutations are represented as single mutations in our instruction design, the exact amount of mutations in each read could be a few mutations more than what the percentage expresses. Figure 16 shows the behavior of UdeAcompress compression ratio as the maximum amount of mutations per read is increased.
As expected, the best performance is achieved with fewer mutations per read. The difference between both compressors tends to decrease as the number of mutations is increased. A very small percentage of mutations [0%–2%] could be considered unrealistic in practice, since it refers to almost exacts alignments which are not likely to happen when handling reads of length 1024. But, even in the very unfavorable scenario of a maximum 25% of mutations per read, UdeACompress compresses over 6× more than Kpath. Finally, it can be noted that the range of the compression ratio for a 70× coverage and typical percentages of mutations (8%-10%) can be estimated between 70× and 100×.
In a scenario that included unmapped reads, UdeACompress would still compress better than the rest of available software. When sequencing technologies for longer reads arrive, it must be studied the real impact that longer reads could have in the amount distribution of mutations.
Conclusions
The amount and importance of genomic data will continue to increase in the near future, hence the need to create strategies for its efficient compression and later storage or transmission. A multi-technique scheme for referential compression of FASTQ files has been proposed here. The core algorithms of the referential compressor (alignment and encoding) have been implemented and tested against the best programs available in the state of the art, evaluating its performance in the compression of FASTQ files and in the compression of read sequences only.
When compared to the other available alignment-based referential compressor, UdeACompress had similar or better compression ratios, producing files 14% smaller and decompressing
Most of the current tools have been designed for the compression of FASTQ files with short reads, a common property of the dataset used for the tests presented here. Nevertheless, in UdeACompress, we envisioned a scenario where reads are longer, which is a clear trend in sequencing technology. Using simulated data, we evaluated many scenarios of long reads compression. In all of such tests, UdeACompress achieved high compression ratios, even under unfavorable conditions. The additional mutations and matchings introduced in UdeACompress seemed to have a positive impact in the final compression, but this still needs to be explored as well.
Compression of quality scores and unmapped reads has a great impact in the compression of a FASTQ file, so specialized strategies for their compression need to be developed as well if high compression ratios are to be achieved. In addition, different strategies for compressing the identifiers must be tested, since the re-arrangement of the reads performed by UdeACompress could handicap the delta encoding that was used to compress them. It should be explored if combining the current low-level compressor with another compression strategy as delta encoding or Markovian models could result in a better harnessing of the redundancy among the read sequences.
The speed of UdeACompress is sensitive not only to the length and number of reads in the FASTQ input file, but also to the size of the reference, increasing the amount of CPU and memory required. Even when the execution times of UdeACompress were acceptable in comparison to relevant software in the state of the art, our model allowed us to estimate a noticeable superior performance if parallelizing the seeding and encoding functions of UdeACompress. Heterogeneous hardware and mixed approaches like coarse and fine grain parallelism must be considered in this extent. Finally, UdeACompress stands out as an effective alternative for compressing not only FASTQ files, but also the genomic alignment data.
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the Administrative Department of Science, Technology and Innovation of Colombia (COLCIENCIAS), Call 757, Grant BEC17-2-27, and by the University of Antioquia through multiple CODI instruments.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
AG and SI conceived and designed the study and experiments, analyzed the results and wrote the manuscript. AG designed and implemented the encoding algorithm and put together all the blocks of UdeACompress, and run the experiments. JL designed and implemented the alignment algorithm. SI and JEA did a critical review of the manuscript and provided guidance throughout the study.