
Abstract
Bluetongue virus (BTV) is an arbovirus considered as a major threat for the global livestock economy. Since 1999, Tunisia has experienced several incursions of BTV, during which numerous cases of infection and mortality have been reported. However, the geographical origin and epidemiological characteristics of these incursions remained unclear. To understand the evolutionary history of BTV emergence in Tunisia, we extracted from Genbank the segment 6 sequences of 7 BTV strains isolated in Tunisia during the period 2000 to 2017 and blasted them to obtain a final dataset of 67 sequences. We subjected the dataset to a Bayesian phylogeography framework inferring geographical origin and serotype as phylodynamic models. Our results suggest that BTV-2 was first introduced in Tunisia in the 1960s and that since 1990s, the country has witnessed the emergence of other typical and atypical BTV serotypes notably BTV-1, BTV-3 and BTV-Y. The reported serotypes have a diverse geographical origin and have been transmitted to Tunisia from countries in the Mediterranean Basin. Interserotype reassortments have been identified among BTV-1, BTV-2 and BTV-Y. This study has provided new insights on the temporal and geographical origin of BTV in Tunisia, suggesting the contribution of animal trade and environment conditions in virus spread.
Introduction
Bluetongue (BT), is a viral infectious and non contagious disease that infects ruminants, primarily sheep and, to a lesser extent, cattle, such as goats and buffaloes. This vector-borne disease is transmitted and operated by the blood-sucking midges of the genus Culicoides. BT has serious economic consequences, estimated in 2020 to 3 billion US$. 1 The BT causative agent is the bluetongue virus (BTV), a double-stranded RNA virus belonging to Orbivirus genus, with numerous identified serotypes. 2 The viral genome encoding for a total of 10 structural and nonstructural proteins. VP2 and VP5 that compose the virus outer-capsid are the highly variable proteins. 3 Among the prevalent serotypes, serotypes 1 to 24 are considered as classical, whereas the others are considered atypical with temperate clinical signs that primarily affect small ruminants.4,5 The genetic evolution of the virus is based mainly on reassortment that may occur during co-infection of different strains, including wild and vaccine strains. 6 At least 28 of the 35 serotypes harbored low cross-reactivity to vaccines.2,7,8 This led to the adoption of multiple vaccination strategies in regions where multiple serotypes coexist. Moreover, since the early 2000s, the BTV has undergone a significant evolution leading to a nearly worldwide spread.9,10 Indeed, its distribution was initially restricted to subtropical regions, with sporadic penetration into more temperate regions of the world.
In Tunisia, livestock maintenance is strategic toward achieving self-sufficiency for food. In the late 90s, livestock accounted for a significant ratio of agricultural production, representing 40% of total agricultural product. Beef, sheep and goat meat constituted 95% of national production, while 5% came from camels and horses (Tunisian Ministry of Agriculture, 1999). However, the emergence and re-emergence of BTV in Tunisia represents a threat for the livestock sector. Since 1999, death cases among infected ruminants by BTV have been reported. 11 During 2019 to 2020, BTV serological survey demonstrated the circulation of several serotypes notably BTV-1, BTV-2, BTV-3, BTV-4 and BTV-26 in many Tunisian governorates. 12 But, the origin and epidemiological traits of BTV incursions are not yet fully explored. Understanding the evolutionary history of BTV dispersion in Tunisia could be helpful to control the virus emergence.
Considering the above-mentioned matters, the present work aims to explore new aspects of BTV emergence in Tunisia by revealing temporal and spatial dynamics of virus spread as well as investigating ancestral reassortments and hosts.
Material and Methods
VP5 nucleotide sequences collection
Among the BTV genes, the VP5, encoding for the outer capsid protein is characterized by high genetic variability 3 and is therefore a suitable genetic marker to track the evolutionary features of BTV. Therefore, we queried full length nucleotide sequences of segment 6 (S6) encoding the VP5 protein of BTV strains from Genbank. Only 7 sequences of BTV viruses (n = 7) circulating in Tunisia during the period 2000 to 2017 were available in Genbank. These sequences belong to typical serotypes namely BTV-1 (n = 2), BTV-2 (n = 2), BTV-3 (n = 2); whereas atypical serotypes were solely represented by BTVY (n = 1).
For Bayesian phylodynamic analysis purposes, we further included other S6 sequences using the following strategy. First, all the S6 sequences from Tunisia (n = 7) were subjected to the BLASTN tool at NCBI. The resulting list of sequences was used to select the reference sequences based on their epidemiological features where we took one sequence per isolation year, country or region of isolation and BTV serotype and infected host. Considering them as phylogenetically distant from the investigated Tunisian strains, we also discarded from our dataset the S6 sequences presenting less than 67% of similarity. A total of 36 sequences originating from Africa, Europe and America were obtained. We additionally supplied the dataset by nucleotide sequences (n = 24) from North African neighboring countries notably Libya, Algeria and Morocco. Ultimately, we conducted the analysis using a dataset totaling 67 sequences. The overall details are accessible on Table 1 and additional file 1.
Metadata of Bluetongue Virus (BTV) S6 reference sequences (n = 60) used for phylodynamic analysis.

Phylogeographic analyses
MUSCLE software was used to align the selected S6 genes sequences. 13 The obtained multiple sequence alignment was then manually edited using BioEdit software. 14 The sequence alignment was verified for presence of recombinant BTV strains using RDP5. 15 Recombination events are considered significant when the associated Bonferroni-corrected P value is inferior to .05 for 4 or more recombination detection methods implemented in RDP5. 15 Using this criterion, no recombination event was detected in our dataset.
The jModelTest v2.1.10 program 16 identified the General Time-Reversible (GTR + G+I) substitution model as the most appropriate. Phylogenetic analysis was performed using PhyML v3.3.3 17 and the resulting tree was visualized with FigTree v1.4.3. To assess the temporal signal in time-stamped sequences, the Maximum Likelihood (ML) tree was analyzed with root-to-tip regression method using TempEST v.1.5.3 18 and permutation test using BactDating package (https://github.com/xavierdidelot/BactDating). It was reported that TempEST might infer false negative results and using the permutation test could be a good alternative to evaluate the temporal signal of the molecular dataset. 19
To construct the Bayesian maximum-clade-credibility (MCC) phylogenetic trees, the BEAST v1.8.4 20 package was used. To explore the evolutionary history of Bluetongue virus, geographic origin and serotype models were analyzed as a discrete trait diffusion model and their transition rates were estimated using a Bayesian Stochastic Search Variable Selection (BSSVS), with a symmetrical discrete trait substitution model (strict clock assumption). The uncorrelated lognormal relaxed clock model was used in the Bayesian analysis.
The Markov chain Monte Carlo (MCMC) was run under an Extended Bayesian Skyline model for over 300 million generations (sampling every 1000 states). An Effective Sample Size (ESS) was observed for the major parameters. The ESS values were evaluated with TRACER v1.8.4 21 and a cut-off value of 200 (ESS > 200) was used to retain concluding simulations. TreeAnnotator 20 (after removing 10% burn-in) was used to generate the MCC tree.
Ultimately, FigTree v1.4.3 ((http://tree.bio.ed.ac.uk/software/figtree/) was used to visualize the resulting MCC tree. A Posterior probability (pp) threshold higher than .95 was used to assess the tree node robustness. To study the spatio-temporal diffusion of circulating BTV, a Keyhole Markup Language (KML) file was generated from the MCC tree with SPREAD3 22 and then visualized with Google Earth Pro (https://www.google.com/earth/versions/, accessed on 5 December 2021). SPREAD3 was also used to calculate the Bayes factors (BF) for pairwise diffusion rate between sites. Bayes Factors > 3 were considered as statistically significant.
Sampling bias assessment
Sensitivity analysis was performed using the “randomized tip swap” approach 23 to assess the sampling bias for each discrete trait diffusion model (geographic origin and serotype). The XML files generated with BEAST 1.8.4 were modified by including the tip swap operator in the block of operators and each MCMC was run for 50 million iterations sampling every 1000 states. ESS values superior to 200 were obtained for the estimated parameters. 23 For an unbiased sample, the distribution of Root State Posterior Probabilities (RSPP) should be different between the main and the “tip swap” analyses. The values of transition rates should be similar in both types of analysis. In this case, the large transition rate could indicate that the obtained results are under the influence of the sampling scheme and not the genetic data. 23
Results
Phylogeography history of BTV outbreaks in Tunisia
To evaluate the temporal signal of our dataset, we performed the root-to-tip regression method and the permutation test. Based on the root-to-tip method, The R 2 value obtained by TempEST was near to null. However, the permutation test revealed a positive correlation with an R 2 = .14 and P value = 5.2 ×10−3 (Figure S1).
Using the S6 sequences and the Bayesian phylogeography framework, we reconstructed a time scaled MCC tree inferring the ancestral geographic origin of BTV for each branch. As shown by the MCC tree (Figure 1), the Time for Most Recent Common Ancestor (TMRCA) estimation revealed that BTV first appeared in Tunisia in 1966 (95% HPD : 1954-1978) (pp = 0.99) and this was marked by the introduction of strains similar to those of BTV-2 isolated in the country in 2000. Thereafter, other BTV outbreaks occurred probably in 1993 (95% HPD : 1985-2004) (pp ≈ 1), 2004 (95% HPD : 2002-2005) (pp = 0.51) and 2009 (95% HPD : 2004-2013) (pp ≈ 1), most likely due to the circulation of respectively BTV-Y, BTV-1 and BTV-3 in Tunisia. As shown by the MCC tree the direct geographic origin of these serotypes is diverse; including countries in Central Africa (Cameroon), North Africa (Morocco) and the Middle East (Israel) (Figure 1). We also noticed that Cameroon is likely to be the most recent origin for BTV-2 Tunisian isolates (pp = 0.99). Our analysis also showed that BTV-Y and BTV-1 serotypes have probably circulated in Morocco before being introduced in Tunisia (pp = 0.99 and 0.51 respectively) and BTV-3 strains are possibly originating from Israel (pp ≈ 1) (Figure 1).

Bayesian time-scaled MCC tree with geographic origin assignment of Bluetongue virus (BTV). TMRCA, their 95% highest posterior density interval (HPD) and the posterior probabilities (pp) are mentioned on the tree nodes for the Tunisian BTV clades. Blue bars indicate 95% HPD confidence intervals of the node heights. Tree nodes are sized according to posterior probabilities. Branch colors indicate the ancestral geographic origin of the descent nodes.
The spatial and temporal dynamics of the BTV dispersal process suggest that strains from Tunisia are historically rooted in Cyprus and were transmitted to South Africa in the 1940s (Figure 2). During the 60s, these strains emerged from South Africa to Guatemala in Central America and subsequently to other African countries, notably Cameroon and Nigeria. The 70s period has witnessed the first diffusion of BTV in Tunisia, from Cameroon. From the mid-2000s until the 2010s, other BTV serotypes spread from Morocco, Nigeria and Israel to Tunisia (Figure 2). The phylogeographic analysis also revealed that since the late 90s, Tunisia has become a diffusion point of BTV toward the surrounding Mediterranean countries such as Morocco, Libya, Israel and Italy (Figure 2).
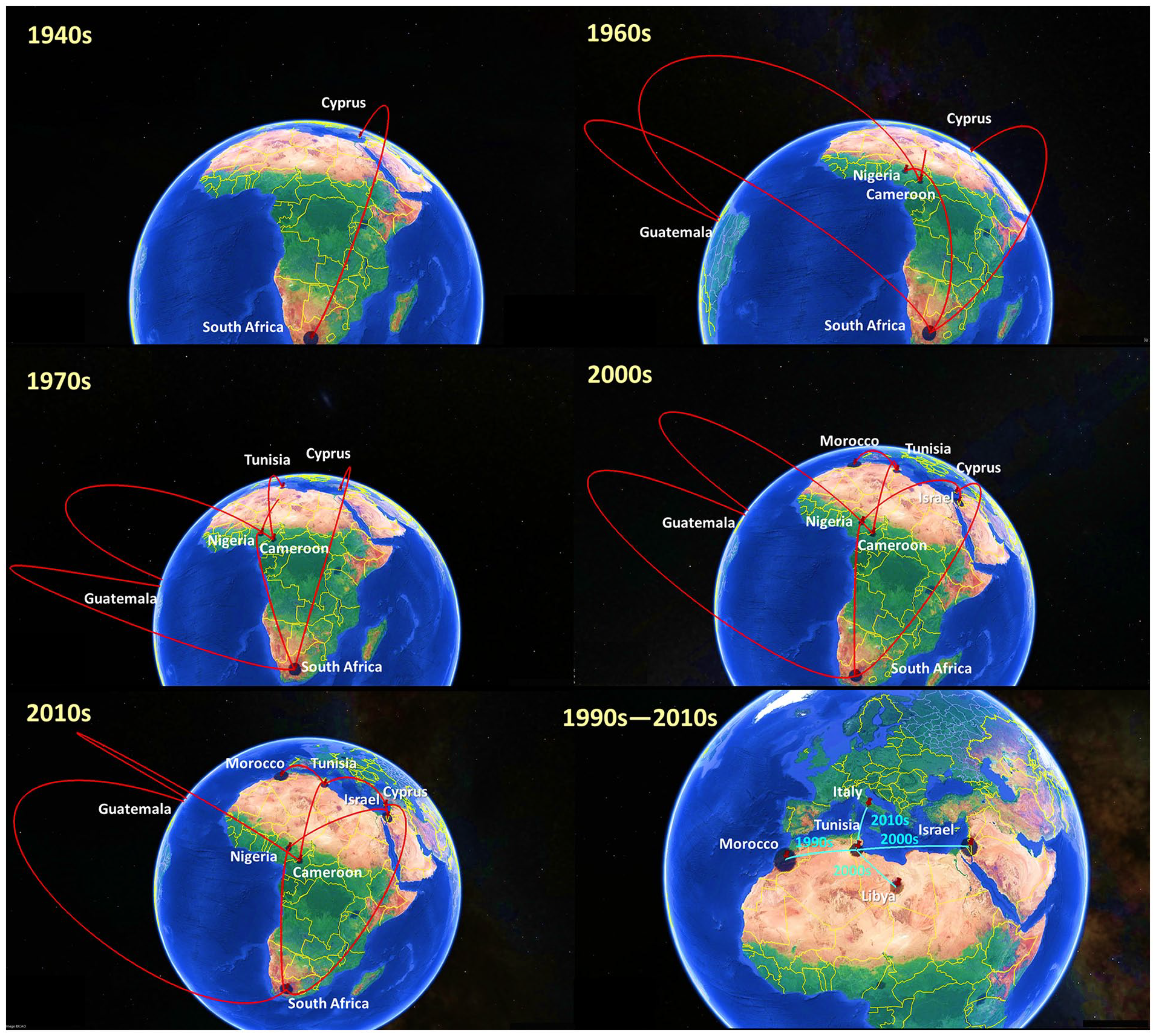
Spatial and temporal dynamics of BTV strains circulating in Tunisia. Red lines: BTV transmission from other countries to Tunisia. Cyan lines: BTV transmission from Tunisia to other countries. The map is created by using Google Earth Pro.
The Bayesian Stochastic Search Variable Selection (BSSVS) approach statistically validated 6 transitions among the 10 identified routes (Figure 3). The transition between Morocco and Tunisia showed the highest BF (BF = 970.78) indicating a decisive support (BF > 100) for this dispersal route. Substantial statistical BF support (BF > 30) was also observed for the transitions Tunisia - Italy (BF = 93.74), Israel–Tunisia (BF = 37.19) and Cyprus–South Africa (BF = 32.17). The routes linking South Africa to Nigeria (BF = 3.48) and Nigeria to Israel (BF = 3.30) presented significant support (BF > 3). Non-significant BTV dispersal routes (BF < 3) were those of South Africa–Guatemala (BF = 1.89), Guatemala–Cameroon (BF = 2.40), Cameroon–Tunisia (BF = 2.56) and Tunisia–Libya (BF = 1.15) (Figure 3). The latter observation nuances the possibility that Cameroon could be the direct source for the first BTV emergence in Tunisia.

Chord diagram of BF evaluating the statistical significance of BTV transitions between countries. Chord width is proportional to the support levels of BF. BF > 100: decisive support; BF > 30: very strong support; BF > 3: substantial support; BF < 3: unsupported transition.
Serotype phylodynamic analysis
The MCC tree reflecting the serotype model showed that the S6 segment of BTV-1, BTV-2, BTV-3 and BTV-Y detected in Tunisia originated from a multiple reassortment of either inter or intra-serotype (Figure 4A). The inter-serotype reassortment has been observed mainly from the 1970s until the 2000s (Figure 4B). Since then, the genetic diversity has regressed where the intra-serotype reassortment became dominant (Figure 4B and C).

Serotype phylodynamics of BTV isolated from Tunisia: (A) Bayesian time-scaled MCC tree of the serotype trait; Blue bars indicate 95% highest posterior density interval (HPD) of the node heights; Tree nodes are sized according to posterior probabilities; Branch colors indicate the ancestral geographic origin of the descent nodes, (B) Bayesian skyline plot showing the effective viral population size through time; Black solid lines are median estimates of NeT (effective viral population size over time); blue shading represents the 95% confidence interval of NeT, and (C) frequency of inter-serotype reassortments over the time.
Only the reassortments of BTV-3 with BTV-14 (BF = 2036.74), BTV-2 with BTV-1 (BF = 5.59) as well as BTV-4 with BTV-Y (BF = 9.34) were statistically supported (BF > 3). The remaining reassortments BTV14–BTV2 and BTV2–BTV4 were not statistically significant as they presented a low BF equal to 1.08 and 1.93 respectively (Figure 5).

Chord diagram of BF evaluating the statistical significance of BTV inter-serotype reassortments. Chord width is proportional to the support levels of BF. BF > 100: decisive support; BF > 3: substantial support; BF < 3: unsupported transition.
Sampling bias
The sensitivity analysis showed that the distribution of root state posterior probabilities (RSPP) between the main and the tip swap analyses was different for geographic origin models. Indeed, Cyprus (RSPP = 0.75) and Tunisia (RSPP = 0.27) were the ancestral root states in main and tip swap analyses respectively (Figure S2A). When we compared the transition rates obtained in these analyses, we noticed that the transitions between countries (n = 9) identified in the spatiotemporal dynamics possess similar rates or the difference is less than one transition (Table S1). This indicates that our dataset selection is unbiased. For the serotype model, the root sate posterior probability construction showed a different distribution between the main and the tip swap analyses. The serotype 1 (BTV-1) was estimated as the ancestral root state (RSPP = 0.97) by the tip swap approach, whereas the observed result indicated that BTV-3 is the ancestral root state (RSPP = 0.99) (Figure S2B). All the transition rates of the identified reassortments were similar and estimated to be different of less than one transition suggesting the absence of sampling bias for the selected dataset in the serotype model (Table S1).
Discussion
Bluetongue virus disease is characterized by a large distribution in the world and causes significant economic losses by affecting the livestock breeding sector. 1 In this study, we used a Bayesian phylodynamic framework to investigate the historical origins of BTV outbreaks in Tunisia by analyzing the temporal and the geographical origins as well as the serotype and the host models.
We identified the first introduction and diffusion of BTV in Tunisia as occurring in the 1960s to 1970s period. However, the first BTV incursion in Tunisia was recorded in the 1990s as reported by Hammami and collaborators. 11 In fact, after the 1950s, the virus spread widely and became prevalent in many countries from different continents.1,24,25 North Africa, where the virus was reported for the first time in 1956 in Morocco, 11 is identified in our study as the main source of BTV outbreaks in Tunisia. Consequently, the latter finding supports that the spread of livestock infection by BTV in Tunisia in that period is very likely and here we provide evidence about earlier circulation of BTV in Tunisia.
Our phylogeographic analysis demonstrated that the 1990s to 2010s period was characterized by intense BTV circulation between Tunisia and other Mediterranean countries. This could be explained by the progress of the ruminant breeding sector since the mid-1980s. It was reported that the proportion of sheep within the Tunisian livestock has increased during this period where they represented 83% of the small ruminants in 2007 against only 57% in 1950. 26 According to the study of Sana et al, 12 the risk of BTV infection in Tunisia increases when it is associated with a large flock size. Al khamis and his collaborators suggested that the Mediterranean basin might be considered as a hotspot for BTV outbreaks, incriminating the role of the climatic conditions and the high abundance of Culicoides species in the virus’ maintenance and emergence. 10 This assumption was corroborated by previous findings demonstrating that the epidemic situation of BTV in Tunisia could be related to a moderate rainfall as well as a hot temperature between 32°C and 34°C. 27 The wind-transport events might also be considered as another contributing factor explaining the virus circulation between the countries surrounding the Mediterranean Basin. Indeed, Rajko-Nenow et al and his collaborators 28 hypothesized that the wind-transport events during 2006 played a role in the introduction of BTV8 infected Culicoides in the Larnaca region of Cyprus from Syria, Lebanon and Israel.
The transmission routes between Tunisia and the neighboring countries (Libya and Algeria) were not identified and supported statistically. Considering the well recognized livestock movements, smuggling and traffic on the borders of the neighboring countries,12,29 this finding is unexpected. The low number of sequences originating from these countries in the public repositories might explain this finding. 12 Further sequencing efforts and reporting would reveal these routes. Interestingly, the first report of atypical serotype BTVY (BTV-Y TUN2017) in Tunisia was in 2017 where the virus was isolated from healthy sheep imported from Libya. 30 However, our results suggest that this atypical serotype has been introduced to Tunisia, earlier and more specifically since 1993 after circulation in Morocco. This result also assumes an earlier circulation of atypical BTV serotype in North Africa. In fact, the atypical serotypes have the ability to be directly transmitted 4 and could infect the small ruminants for years5,31 without expressing clinical signs, 32 hence, these particular features make their diagnosis difficult.
In addition, our phylodynamic analyses suggest that BTV-1, BTV-2 and BTV-Y were transmitted to Tunisia after being subjected to multiple inter-serotype reassortments, especially prior to the 2000s decade. The limitation of inter-serotype reassortment events observed after the 1990s could be justified by the successful application of the recombinant vaccination campaigns against BTV. Such campaigns provide a cross-serotype protection, which has the effect of reducing the BTV genetic diversity worldwide.33,34 Furthermore, the ancestral reassortment events have the ability to enhance replication aptitude of BTV strains in vector Culicoides and to increase transmission efficiency to sheep. 35 Thus, the inter-serotype reassortments identified among BTV-1 and BTV-2 could justify their impactful circulation in Tunisia during 2000-2011, where a massive vaccination strategy of Tunisian sheep livestock has been established since 1999 using live and attenuated vaccines against BTV-1 and BTV-2.11,12 The sensitivity analysis suggests that the ancestral construction of evolutionary history of BTV in Tunisia is based on the genetic data and not the sampling bias impact. We found that some statistically unsupported transitions are unbiased; thus suggesting that these countries or serotypes could not be directly connected and their relationships could be ensured by virus reassortments with other unsampled BTV strains.
Here, we used a self-submitted genetic data due to the lack of a BTV active surveillance programs especially in Tunisia and other countries of Maghreb (eg, Libya and Algeria) and this could present a limitation of our study. Despite the ability of the sensitivity analysis to detect the oversampling impact on our phylodynamic results, the investigation of bias from underreporting is difficult. As discussed before, more BTV sequences from Algeria and Libya could reveal the virus transmission from these neighboring countries to Tunisia. Continued monitoring and sequencing of BTV would certainly enhance future phylodynamic analysis. The host model was subjected to the Bayesian phylodynamic analysis in this study, but the sensitivity analysis indicated the presence of sampling bias in the dataset related to this model. Thus, we have decided to not include it in our study. Furthermore, the data provided in this study seems to be insufficient to investigate in detail the cross-species transmission of BTV in Tunisia. Therefore, sampling the virus from different ruminant species is needed.
The present report is the first phylodynamic study providing a historical overview about BTV outbreaks in Tunisia through origin investigation and transmission quantification. Here, we found new evidence about the temporal and spatial origin of BTV epidemics in Tunisia. The multiple transmission routes of the virus could explain the diversity of BTV serotypes in Tunisia. Animal trade, cross-border traffic and environmental factors have a high contribution in the epidemic situation of BTV in Tunisia. Such study is necessary to understand the evolutionary dynamics of BTV and hence helps in monitoring the virus spread in the country regarding its high evolutionary rates and the emergence of alarming emergent serotypes.
Supplemental Material
sj-docx-1-evb-10.1177_11769343231212266 – Supplemental material for Retrospective Phylodynamic and Phylogeographic Analysis of the Bluetongue Virus in Tunisia
Supplemental material, sj-docx-1-evb-10.1177_11769343231212266 for Retrospective Phylodynamic and Phylogeographic Analysis of the Bluetongue Virus in Tunisia by Oussema Souiai, Marwa Arbi, Mariem Hanachi, Ameny Sallami, Imen Larbi, Melek Chaouch, Emna Harigua-Souiai and Alia Benkahla in Evolutionary Bioinformatics
Supplemental Material
sj-eps-3-evb-10.1177_11769343231212266 – Supplemental material for Retrospective Phylodynamic and Phylogeographic Analysis of the Bluetongue Virus in Tunisia
Supplemental material, sj-eps-3-evb-10.1177_11769343231212266 for Retrospective Phylodynamic and Phylogeographic Analysis of the Bluetongue Virus in Tunisia by Oussema Souiai, Marwa Arbi, Mariem Hanachi, Ameny Sallami, Imen Larbi, Melek Chaouch, Emna Harigua-Souiai and Alia Benkahla in Evolutionary Bioinformatics
Supplemental Material
sj-eps-4-evb-10.1177_11769343231212266 – Supplemental material for Retrospective Phylodynamic and Phylogeographic Analysis of the Bluetongue Virus in Tunisia
Supplemental material, sj-eps-4-evb-10.1177_11769343231212266 for Retrospective Phylodynamic and Phylogeographic Analysis of the Bluetongue Virus in Tunisia by Oussema Souiai, Marwa Arbi, Mariem Hanachi, Ameny Sallami, Imen Larbi, Melek Chaouch, Emna Harigua-Souiai and Alia Benkahla in Evolutionary Bioinformatics
Supplemental Material
sj-xlsx-2-evb-10.1177_11769343231212266 – Supplemental material for Retrospective Phylodynamic and Phylogeographic Analysis of the Bluetongue Virus in Tunisia
Supplemental material, sj-xlsx-2-evb-10.1177_11769343231212266 for Retrospective Phylodynamic and Phylogeographic Analysis of the Bluetongue Virus in Tunisia by Oussema Souiai, Marwa Arbi, Mariem Hanachi, Ameny Sallami, Imen Larbi, Melek Chaouch, Emna Harigua-Souiai and Alia Benkahla in Evolutionary Bioinformatics
Footnotes
Author Contributions
Conceptualization, O.S, A.M, I.L and ABK; data curation O.S, MC, E.HS and M.A.; formal analysis, O.S, M.A.; investigation, O.S, EHS and A.M.; methodology, O.S, MH, A.S and M.A.; supervision, O.S and A.BK; writing—review and editing, E.HS, M.C, A.BK, M.H, I.L, O.S. and M.A. All authors have read and agreed to the published version of the manuscript.
Funding:
The author received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.