
Abstract
Microbial community is ubiquitous in nature, which has a great impact on the living environment and human health. All these effects of microbial communities on the environment and their hosts are often referred to as the functions of these communities, which depend largely on the composition of the communities. The study of microbial higher-order module can help us understand the dynamic development and evolution process of microbial community and explore community function. Considering that traditional clustering methods depend on the number of clusters or the influence of data that does not belong to any cluster, this paper proposes a hypergraph clustering algorithm based on game theory to mine the microbial high-order interaction module (HCGI), and the hypergraph clustering problem naturally turns into a clustering game problem, the partition of network modules is transformed into finding the critical point of evolutionary stability strategy (ESS). The experimental results show HCGI does not depend on the number of classes, and can get more conservative and better quality microbial clustering module, which provides reference for researchers and saves time and cost. The source code of HCGI in this paper can be downloaded from https://github.com/ylm0505/HCGI.
Keywords
Introduction
There are many kinds of microorganisms, which exist in human, plant, soil, ocean and other environments. Microbial community can be defined as a collection of microorganisms that exist at the same time and may interact with each other. They exist in a certain spatiotemporal habitat. The term microbial community was first coined by Lederberg and McCray 1 By definition, microbial community is ubiquitous in nature, which has a huge impact on the living environment and human health.2-4 All these impacts of microbial community on the environment and its host are usually called the functions of these communities, which largely depend on the composition of the community, that is to say, on the species and quantity of the existing species. In essence, the interaction between species involves multiple species, which often occurs in a high-order combination way, that is, the interaction between two species is regulated by one or more other species, 5 and the high-order interaction often dominates the function of microbial community, for example, higher-order functional interaction dominates the hydrolysis rate of amylase in biological communities. 6
In order to quantify how the interaction between species affects the overall community function, two interactions between species were systematically measured through experiments, such as cross feeding between E. coli growth nutrient bacteria7,8 and naturally isolated bacteria.9,10 Other studies infer the interaction between species by measuring the population dynamics of a complex community.11-14 Some studies mainly focus on the interaction between two species and the ability of two interaction models to predict network functions.15,16 Although the interaction between the two is enough to describe the important impact of ecology in some cases, some studies have emphasized the need to merge higher-order relationships, mainly because of their role in population dynamics.5,17,18
Tsai et al 19 developed a rule-based microbial network (RMN) algorithm, which uses a triple relationship similar to the activation inhibitor target model to build a gene regulatory network. Based on this principle, RMN algorithm was developed to describe the relationship between every three microorganisms as a cooperation competition target. Guo and Boedicker 20 found that the interaction between species has an impact on the activity of the whole community, and studied the interaction network of four microbial communities through theoretical models. The results show that the interaction between multiple species determines the overall metabolic rate of the microbial community. 20 Bairey et al 5 found that the high-order interaction has a lower bound to diversity, while the pairwise interaction exerts an upper bound. When the pairwise interaction is dominant and the number of species increases beyond the threshold, the feasibility decreases. On the contrary, when the four-way interaction is dominant, the community becomes sensitive to species removal. In the case of mixing, when the pairwise interaction and the high-order interaction are dominant, the community becomes sensitive to species removal. When combined, the intermediate species is the most feasible, and the removal and addition of species will destroy the stability of the community. Therefore, the combination of high-order interaction and pairwise interaction determines the most stable range of diversity. These findings emphasize the importance of high-order species interaction in determining the diversity of natural ecosystems. In order to study the contribution of pairwise interaction and higher-order interaction to community function, Alicia Inspired by the complex genetic interaction, etc. Used the ecological function landscape to separate and quantify the single, paired and higher-order interactions in the microbial community function, screened the experimental results through the enzyme dynamics theory, and found that the higher-order functional interaction dominated the hydrolysis rate of amylase in our community. 6 That is to say, it is very important to build a meaningful microbial network to study the high-order interaction between microorganisms.
From the perspective of modules, we can intuitively understand the interaction information and the contribution of network modules to the overall network, so as to study the possible functions of modules formed by specific microorganisms in the microbial community. Girvan and Newman 21 proposed a method to detect the community structure, based on the centrality index to find the community boundary, and applied it to study many different social networks and biological networks, focusing on the least central edge and the most central edge between communities, not by adding the strongest edge to an initial empty vertex set to build the community, but by gradually removing edges from the original graph to build a community can help you understand the composition of other complex datasets.
Copeland et al 22 used the network analysis method to study the relationship between the abundance of each genus in the leaf microbial community, determined two highly connected clusters, and found functional groups. Aleksej considering the possibility of high-order interaction involving more than two species, extended the concept of binary symbiosis to at most four species at the same time, and proposed a model for large microbial communities. The experimental results showed that although there was obvious resource competition at the level of the whole community, microbial communities still had interdependent metabolic groups, and they occur repeatedly in different habitats, emphasizing that metabolic dependence is the main driving force of species symbiosis, and suggesting that the cooperative group is a recurring module in the microbial community structure. 23 That is to say, the study of higher-order microbial modules can help us to understand the dynamic development and evolution process of microbial communities, help us to understand the functional composition of communities, and predict the potential functionality of community modules.
The traditional clustering problem only studies the pairing relationship between two microorganisms. Most current algorithms assume that there are paired similarities between microorganisms, but many microorganisms in the real environment involve more complex and multipath relationships. In such networks of high order relationships, modeling pairs only results in the loss of information in the original data. The graph model of pairwise relations does not consider higher order information, and cannot represent the whole ecological network. Microorganisms diversity depends heavily on the stabilization of higher-order interactions. It is important to study the higher- order interaction of microorganisms to understand the diversity and stability of microbial community. Hypergraph clustering refers to the process of extracting the most consistent group from a group of objects by using high-order (rather than pairwise) similarity. Therefore, hypergraph clustering is applied to study the high-order interaction network module of microorganisms.
In this paper, first, the microbial abundance data were processed, from the perspective of logical relationship, the information entropy is used to determine the high-order interaction relationship of microorganisms, and then the high-order relationship hypergraph of microorganisms is constructed. Then, a kind of microbial logical relationship is selected for further study, and game theory is introduced to transform hypergraph clustering problem into clustering game problem. Finally, we use Baum-Eagon theorem to generate the clustering module with iterative elimination strategy until the remaining objects cannot form a high-order relationship.
Materials and Methods
The dataset
In this paper, the available microbial abundance data of healthy humans based on 16s rRNA comes from the human microbiome project (HMP), which is V13 high quality files processed with the mothur software package. The data covered 18 of the 5 main body parts. Data source web site is http://hmpdacc.org/HMMCP/.
Hypergraph clustering based on game theory
In the previous work, we used to find the maximum degree of modularity to determine the number of clusters before the premise of clustering. 24 In this paper, we studied the hypergraph clustering problem from the perspective of game theory to analyze the high-order module analysis of microorganisms, which can automatically generate clusters without specifying the number of clusters.
In the process of biological evolution, animal adaptability is formed in the interaction with their living environment. In the competition, animals finally choose the evolutionary stability strategy (ESS), which is adopted by most members of the population and will not be eroded by other strategies. In our framework, the clustering problem is seen as a non-cooperative game between multiple microorganisms, and an important concept in game theory is equilibrium, in which the performance of each player in a population does not exceed the population average return when Nash equilibrium is reached. Bulo et al 25 proved that the problem of finding these equilibrium points (clusters) is equivalent to solving a polynomial optimization problem with linear constraints, and used an algorithm based on Baum-Eagon inequality to solve this problem. We apply it to the analysis of the microbial high order module in this paper.
Game theory
A game can be represented as
Given the set of all possible states of the population D:

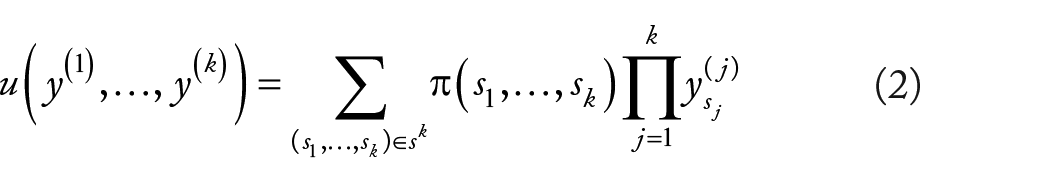
This function insensitive to order of inputs. We suppose that randomly pick people from
Indeed,
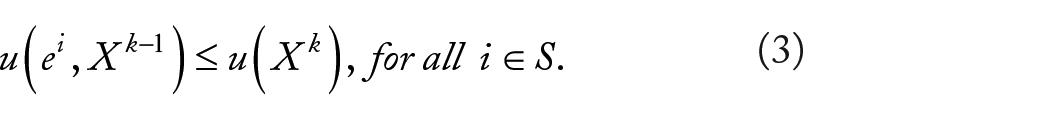
Hypergraph clustering based on game theory

where
Given a population
The problem of extracting ESSs of our hypergraph clustering game can be transformed into the problem of finding strict local solutions of (5). Consider the following nonlinear optimization problem:

By the Karush-Kuhn-Tucker (KKT) conditions there exist
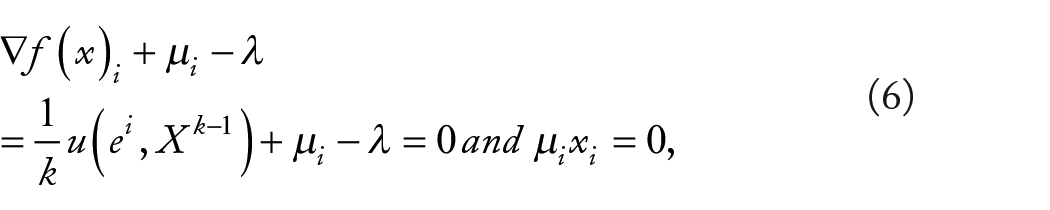
The hypergraph clustering problem of extracting ESSs (evolutionary stability) can be transformed into finding a strict solution of (5). Baum and Eagon 26 are introduced to find ESSs. 21 For maximizing polynomial function in probability domain, The Baum-Eagon inequality is an effective iterative method.

Then
Using the above method, we iteratively find a cluster and remove it from the object set, then repeat the process on the rest of the objects until the remaining nodes cannot form a hyperedge after eliminating the nodes in the cluster.
The data processing
The traditional microbial network construction is based on graph-theory. In this paper, eight logical relationships (see Table 1) among microorganisms are calculated based on the method of entropy. Bowers et al 27 proposed a computational method based on genomic data to identify the detailed relationships between proteins. Based on information entropy, Bowers et al calculates correlations between proteins and identifies eight logical relationships, which reveals many previously unknown higher-order relationships. We applied it to construct microbial higher-order networks.
Description of the logical relationship between microbes and the total number of microbes present in the human body.

Table 1 describes logical relationships among microorganisms and the total number of each logical relationship in human body based on the selected dataset. Venn diagram and related logic statements in the Table 1 illustrate eight different logical relationships, which represent different co-occurrence relationships among three Microorganisms. In other words, these describe the possible dependence of the existence of C on the existence of A and B. It can be seen from Table 1 that the most common logical relationships among microorganisms in human body are type1, type3 and type7. The discussion of logical relations with less relations is of little significance. We can choose type1, type2 or type3, but the number of type 3 relationships is large, and the interaction between microorganisms is more complex, which is not easy to analyze, so type 1 is finally selected.
Figure 1 shows the calculation process of high-order logical relationship of microorganisms.
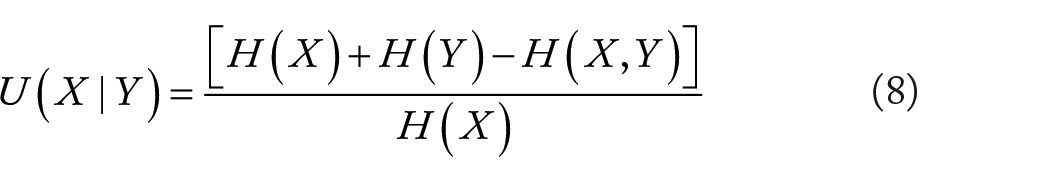

The calculation process of high-order logical relationship of microorganisms. (A) Extract microbial abundance data from open source websites converted into 0 to 1 microbial abundance matrix. (B) Calculate the number of 8 high order logical relationships in the presence of microorganisms. (C) Select type1. (D) Calculate the uncertainty coefficient
The maximum fraction value of uncertainty coefficient obtained by the triplet is taken as the initial weight of the hyperdge.
Construction of weight matrix s(e)
Weight each hyperedge with a positive value, we can defined it with
M={


Trace (﹒) is the average measure of characteristic variance. The smaller trace (﹒) is, the smaller the characteristic variance is and the higher the tightness within the class is. So, We can construct the weight of the hyperedge in the following way:

Where
Results and Analysis
Evaluation index
Joint entropy
The joint entropy (JE) is a measurement uncertainty evaluation method to measure the uncertainty related to a set of variables. Take JE as a standard of measuring the information contained in a cluster. Joint entropy is a measure of uncertainty associated with a set of variables, which is used to measure the amount of information contained in the same cluster of microorganisms. The smaller the joint entropy, the smaller the amount of expression information.

where
Total correlation
Total correlation (TC) 28 is derived from mutual information generalization. Mutual information is an information measure in information theory, which can be regarded as the information contained by one random variable about another random variable. TC is an effective method to measure the independence between a set of microbes and to evaluate the degree of microbial interdependence within the same module. When the total correlation is close to 0, the microorganisms in the module are statistically independent. The lower the total correlation, the lower the interdependence, thus ensuring the quality of the high-order interaction in the module.

Comparison of algorithms
In this paper, the joint entropy, total correlation and modularity are taken as the indexes to evaluate the clustering performance of the algorithm, the hyperspectral clustering based on game theory and intra-class scatter matrix (HCGI) and hypergraph clustering based on intra-class scatter matrix (HCIS). 24 They were applied to cluster analysis and comparison of microorganisms in 18 parts of human body. Based on the clustering results, the JE and TC of each cluster were calculated. we take the sum of JE of the subclusters and the sum of TC as an final index to evaluate the performance of an algorithm. The sum of JE represents the total expression information of the microbial cluster, and the sum of TC represents the total dependence within the microbial community.
We selected the mid vagina where the clustering effect did not change much after the addition of intra-class scatter matrix and intestines tract where HCIS did not work well in to compare and analyze in the previous work. 24
Tables 2 and 3 show that cluster analysis and comparison of mid vagina and intestines tract based on type 1. We can see that the two methods end up with different clustering numbers on the same body part. From Table 2, we can observe that the sum of JE generated by HCGI algorithm is higher than that of HCIS, while the sum of TC is lower than that of HCIS, which indicates that the results generated by HCGI clustering algorithm express more total information, and the correlation between the generated clusters is weaker. But in Table 3, the correlation between the generated clusters by HCIS is weaker, this is because the nodes removed by the two methods are not consistent, so it is not good to use the above evaluation indicators to evaluate the clustering performance of the two methods. However, according to joint entropy and total correlation of each cluster, we can judge the amount of expressed information of the cluster and the connection strength between nodes. Next, through the case analysis of mid vagina, we can visualize the clustering results of mid vagina, and specifically analyze the advantages and disadvantages of two methods.
The clustering analysis and comparison of mid vagina based on type1.

The clustering analysis and comparison of intestines tract based on type1.

Visual analysis
Cytoscape 29 has proven to be a high-level platform for visualization and analysis of biological networks. We used cytoscape to analyze the clustering results of HCGI on mid vagina and find the differences between nodes within each cluster.
Figure 3 shows the clustering results of HCGI algorithm on mid vagina. The nodes in the box inside the black dotted box belong to the same cluster. HCGI algorithm divides the nodes on mid vagina into 7 clusters. The blue nodes that are not framed by the black dotted line do not belong to any of the clusters. Figure 2 shows the clustering results of HCIS on mid vagina. HCIS divides the nodes on mid vagina into 5 clusters. In Figure 2, the blue square nodes do not belong to any of the clusters. Compare the clustering results of Figures 2 and 3, we can find that HCGI divides more clusters than HCIS algorithm. However, it can be observed that the cluster generated by HCGI algorithm is contained in the cluster generated by HCIS algorithm. The nodes eliminated by HCGI algorithm are also nodes connected sparsely to the network graph, and the completeness of the higher-order organization is still maintained in the process of iterative elimination. Based on the framework of game theory, we can get better and more conservative microbial modules.

The cluster results of HCIS on mid vagina.

The cluster results of HCGI on mid vagina.
Conclusion
Studying microbial community interactions is challenging because the microorganisms in a community often interact with each other in many ways, not just in pairs. The basic way to detect the biological significance of biological networks is based on the module perspective.
On the basis of hyperedge similarity matrix reconstructed by intra-class scatter matrix in our previous work, this paper introduces game theory and proposes hypergraph clustering based on game theory (HCGI) algorithm for mining microbial higher-order module research. Hypergraph clustering problem were converted to non-cooperative multi-player game clustering problems, and get the optimal clustering module by finding countermeasures balance. There is no need to specify the cluster number in advance, and one module is obtained after each iteration until the remaining object set cannot build the higher-order relationship. The experimental results show that the introduction of game theory makes the clustering module more conservative and the quality of the module better, which indicates that the higher-order module of hypergraph clustering based on game theory is effective.
Future Work
This paper is mainly based on information entropy to build non directional high-order microbial interaction relationship. The interaction relationship in real biological network may be directional relationship, such as metabolic relationship. Therefore, we can analyze and construct the possible high-order relationship between directed microorganisms from the perspective of metabolism.
With the development of time series data, many dynamic models have been used in microbial networks. Dynamic models such as dynamic Bayesian models can be used to construct high-order interaction networks among microorganisms.
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China [61532008, 61872157, 6192008]; the Self-determined Research Funds of CCNU from the Colleges’ Basic Research and Operation of MOE [CCNU19QD003]; and the National Language Commission Key Research Project [ZDI135-61].
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
LY and XS designed and implemented the computing framework. LY and XS analyzed the results and wrote the manuscript. LY, XS, JY, KW, DZ and RX revised the manuscript. LY prepared the computational codes and carried out. All the authors wrote, reviewed and approved the final manuscript.