
Abstract
Self-interacting proteins (SIPs) play crucial roles in biological activities of organisms. Many high-throughput methods can be used to identify SIPs. However, these methods are both time-consuming and expensive. How to develop effective computational approaches for identifying SIPs is a challenging task. In the article, we present a novel computational method called RRN-SIFT, which combines the recurrent neural network (RNN) with scale invariant feature transform (SIFT) to predict SIPs based on protein evolutionary information. The main advantage of the proposed RNN-SIFT model is that it uses SIFT for extracting key feature by exploring the evolutionary information embedded in Position-Specific Iterated BLAST–constructed position-specific scoring matrix and employs an RNN classifier to perform classification based on extracted features. Extensive experiments show that the RRN-SIFT obtained average accuracy of 94.34% and 97.12% on the yeast and human dataset, respectively. We also compared our performance with the back propagation neural network (BPNN), the state-of-the-art support vector machine (SVM), and other existing methods. By comparing with experimental results, the performance of RNN-SIFT is significantly better than that of the BPNN, SVM, and other previous methods in the domain. Therefore, we conclude that the proposed RNN-SIFT model is a useful tool for predicting SIPs, as well to solve other bioinformatics tasks. To facilitate widely studies and encourage future proteomics research, a freely available web server called RNN-SIFT-SIPs was developed at http://219.219.62.123:8888/RNNSIFT/ including the source code and the SIP datasets.
Introduction
Protein-protein interaction (PPI) prediction revealed multiple roles in many important biological activities. However, an interesting related research problem is whether proteins can interact with their partner. Self-interacting proteins (SIPs) is being considered as a special type of PPIs, which refers to more than 2 copies of the protein can interact with each other and are the same copies of the protein and can be represented by the same gene. This might bring about the formation of homo-oligomer problem. Many recent studies have shown that SIPs play a vital role in various cellular physiological functions and the evolution process of protein-protein interaction networks.1,2 Therefore, whether a protein can self-interact for interpretation of its functions is very important. The research on SIPs can provide a better understanding of the regulation of protein function and the molecular mechanisms involved in biological activity and the underlying cellular and genetic disease mechanisms. Many studies have been conducted for the homo-oligomerization that is a vital function for biological activity and plays an essential role in a wide range of biological processes, such as signal transduction, gene expression regulation, enzyme activation, and immune response.3-7 In addition, it has been demonstrated by many previous studies that the diversity function of proteins can be variously extended without increasing the length of genome through SIPs. Self-interacting proteins can also provide some help in improving the protein stability and preventing the protein denaturation by reducing its surface area.8,9 Therefore, it is becoming more important to develop reliable and effective computational approaches based on protein sequences for predicting SIPs.
Also, more research has been devoted to develop computational methods to predict PPIs. Gao et al 10 proposed a novel computational method called RF-AC, which combined the Rondom Forest (RF) classifier with Autocovariance (AC) approach–based position-specific scoring matrix (PSSM). Huang et al 11 presented a new computational approach, which used weighted sparse representation as classifier and employed global encoding as a feature extraction method for predicting PPIs. Pan et al 12 proposed a novel latent Dirichlet allocation-rondom forest (LDA-RF) model for predicting human PPIs based on protein primary sequences, which has strong ability for processing large-scale datasets by using the LDA-RF model. Zhang 13 proposed a novel approach based on protein sequence that used random tree and genetic algorithm for predicting PPIs. Yang et al 14 presented a new approach that used local descriptors to represent protein sequence and employed the k-nearest neighbors for performing classification. Guo et al 15 adopted autocorrelation feature extraction technique for generating feature vectors and used the support vector machine (SVM) classifier to identify PPIs. An et al 16 proposed a classification algorithm of compound kernel function RVM based on gray wolf optimization algorithm and k-fold cross-validation, which fully consider the special features of local and global of PPI position. An et al 17 proposed a feature extraction approach based on local protein sequence PSSM matrix coding and serial multifeature fusion. The method can capture PPI information of continuous and discontinuous for protein sequence by using the local protein sequence PSSM matrix coding; much key feature information–contained protein sequences can be integrated through employing serial multifeature fusion. These methods usually explore the correlational information between protein pairs, such as coevolution, colocalization, and coexpression. However, this information is not enough for predicting SIPs. In addition, the PPI datasets do not contain the PPIs between the same protein partners. For all these reasons, it is not adequate for predicting SIPs by using these computational approaches. In a previous study, Liu et al 18 proposed a method integrating multiple representative known properties to create a prediction mode called as SLIPPER to predict SIPs. As far as we know, a number of recent studies have been reported about PPIs, which may also be related to SIPs.19-24 However, there is obviously a drawback that cannot deal with the proteins not covering the current human interatomic by using these methods. Due to all the reasons presented, the development of efficient computational methods for predicting SIPs is a necessary work.
In the study, we proposed a new computational approach called RRN-SIFT, which combines the recurrent neural network (RNN) with scale invariant feature transform (SIFT) for predicting SIPs based on protein evolutionary information. The proposed method uses SIFT to extract key features from PSSM that is constructed by using the Position-Specific Iterated BLAST (PSI-BLAST) tool and contains protein evolutionary information. The RNN classifier is employed for executing classification based on extracted features. The RRN-SIFT model obtained average accuracy of 94.34% and 97.12% on the yeast and human dataset, respectively. Compared with the back propagation neural network (BPNN), the state-of-the-art SVM, and previous computational models, our method takes full advantage of RNN and SIFT, thereby improving the prediction accuracy. Therefore, the experimental results demonstrated that the proposed RNN-SIFT model is a useful tool for predicting SIPs and is also suitable for other bioinformatics tasks.
Materials and Methods
Dataset
The UniProt database contains 20 199 curated human protein sequences. 25 The PPI datasets can be downloaded from different databases, including DIP, 26 BioGRID, 27 IntAct, 28 InnateDB, 29 and MatrixDB. 30 In the article, we constructed the PPI data that only contain the same 2 interaction protein sequences and whose interaction type was defined as “direct interaction” in relevant databases. As a result, we acquired 2994 human self-interaction protein sequences. To verify the performance of the RNN-SIFT model, we constructed the experimental datasets by using the following 3 steps: (1) the protein sequences with length less than 50 residues and longer than 5000 residues were removed from the whole human proteome; (2) we selected the SIP data to create the positive dataset, which must satisfy with 1 of the following conditions: (a) it has been detected for self-interacting by at least 2 kinds of large-scale experiments or 1 small-scale experiment, (b) the protein has been defined as homo-oligomer (including homodimer and homodimers) in UniProt, and (c) it has been reported by at least 2 publications for self-interacting; and (3) for constructing the negative dataset, we removed all types of SIPs from the whole human proteome (including proteins annotated as “direct interaction” and more extensive “physical association”) and UniProt database. Consequently, we selected 15 938 non-SIPs as negative samples and 1441 SIPs as positives samples for creating the human dataset. 31 In addition, we also used the same strategy to construct the yeast dataset that contains 5511 negative and 710 positive samples. 31
Feature extraction method
Position-specific scoring matrix
Position-specific scoring matrix contains not only the position information but also the evolution information of protein sequence. As a result, the PSSM is used to extract the evolutionary information in the article. Position-Specific Iterated BLAST 32 is used to convert each sequence into a PSSM. Assuming the length of a given protein sequence is L, its PSSM can be expressed as an L × 20 matrix. Figure 1 shows the schematic of a PSSM.

The schematic of a PSSM.
In the artwork, L represents the length of a given sequence, 20 is the number of 20 amino acids, and
Scale invariant feature transform
Scale invariant feature transform is an image descriptor for image-based matching and recognition developed by Lowe.33,34 The original SIFT descriptor was calculated from the image intensities around interesting locations in the image domain which can be named interest points or key points. These interest points are obtained from scale-space extrema of difference of Gaussians (DOG) within a DOG pyramid. Lindeberg35,36 proposed a new method for finding out interest points by using the SIFT approach. This method can be viewed as a variation of a scale-adaptive blob detection approach, where blobs with associated scale levels are detected from scale-space extrema of the scale-normalized Laplacian. The scale-normalized Laplacian is normalized with respect to the scale level in scale space and is defined as
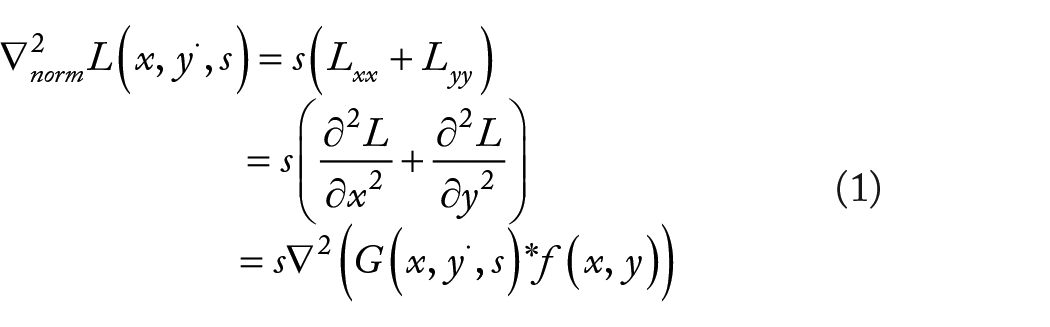
For obtaining the maximum value of the DOG image under different scale magnifications, the smoothed image value of a given original image is convolved with Gaussian kernels of different widths by using the SIFT algorithm, a scale-variable Gaussian function is defined as follows

These Gaussian blurred images are grouped according to their scale magnification, so the number of Gaussian blurred images processed in each group is the same. At this time, the DOG image can be obtained by subtracting 2 adjacent Gaussian blurred images in the same group. The DOG operator constitutes an approximation of the Laplacian operator of different widths, which denotes the standard deviation and the variance of the Gaussian kernel. The DOG operator which constitutes an approximation of the Laplacian operator is defined as follows

Which by the implicit normalization of the DOG responses, as obtained by a self-similar distribution of scale levels

After the DOG image is obtained, the maximum and minimum values can be found and is referred to as key points in the DOG images. To quickly find the key points, each pixel of the DOG image will be compared with 8 pixels around itself and 9 pixels at the same position in the same group of the DOG images at adjacent scales. The maximum and minimum values of these pixels are called key points. As a result, the critical point detection of SIFT algorithm is actually a variant of blob detection, which use Laplacian to compute the maximum value in each magnification space. The Gaussian difference can be approximated as the result of Laplace operator operation. The SIFT employs the concept of “scale space” to capture features at multiple scale levels or image resolutions, which not only increases the number of available features but also makes the method highly tolerant to scale changes.
In the article, we assumed that each PSSM is an image matrix. As a result, we used the SIFT feature extraction method to generate feature vectors and its dimensional is 128. The technology roadmap of the proposed method is shown in Figure 2.

The technology roadmap of the proposed method.
Recurrent neural network
Recurrent neural network is a machine learning method based on deep learning, which is used to solve binary or multiple classification problems. For tasks that involve sequential inputs, such as speech and language, it is often better to use RNNs. RNNs process an input sequence one element at a time, maintaining in their hidden units a “state vector” that implicitly contains information about the history of all the past elements of the sequence. The final output of the RNN model is the classification label of each feature vector.
Recurrent neural network is used to solve the problem that the input training sample is a continuous sequence and the length of the sequence is different, such as the problem based on time series. The basic neural network only establishes weight connections between layers. The biggest difference of RNN is that the weight connections also established between layers of neurons.37-39 The structure of RNN is as follows:
It can be seen from Figure 3 that the output of RNN at any moment is related to the current input and the previous output. RNN’s forward propagation is a combination of multiplication, addition, and set operations. It is well known that t moment of a given ordered sequence will lead to computation of the hidden layer t times. The current state of hidden layer

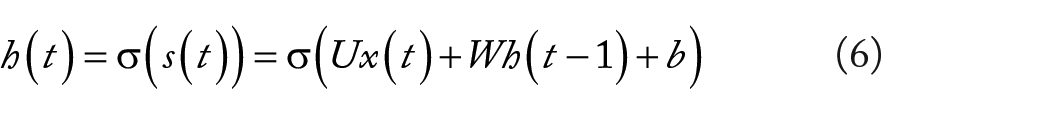
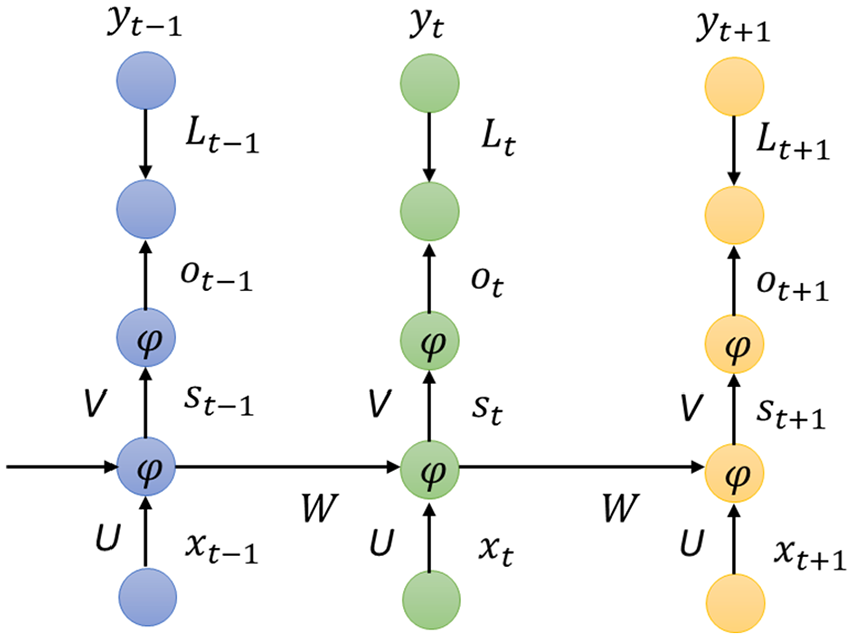
The structure of RNN.
where

The softmax function can be used to perform classification and output the final prediction probability value, which is shown as follow

Here, the loss function of

The loss function (global loss) of the RNN model at all moments N can be expressed as follows

The gradient of 3 parameters U, V, and W of the global loss can be defined as follows
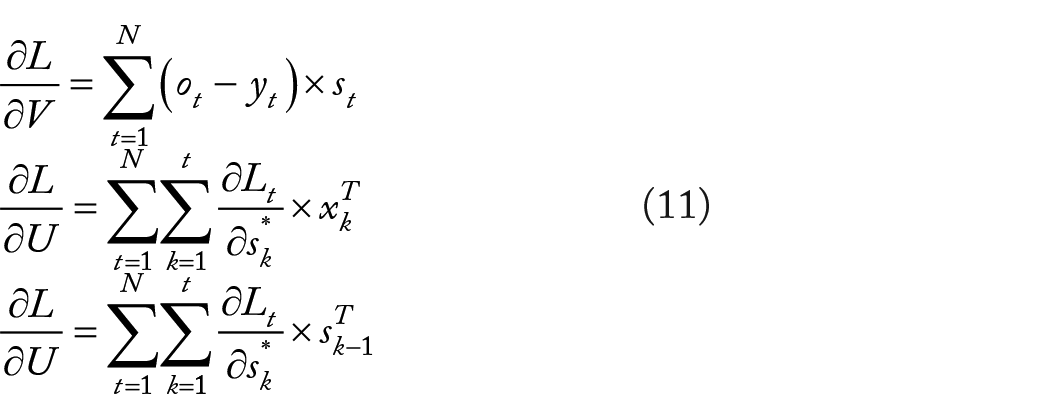
The most commonly used method for optimization problems is the gradient descent. In the article, the gradient update for the 3 parameters can be expressed as follows

The major advantage of the RNN model in learning nonlinear sequential data is well known and has been used in language modeling and sequential labeling. In consideration of SIPs dataset is also a kind of nonlinear sequence data, so we used the RNN model to predict SIPs in the study. The prediction flowchart of RNN-SIFT model is displayed in Figure 4.

The prediction flowchart of RNN-SIFT.
Performance evaluation
In the article, we employed the following measures to assess the performance of RNN-SIFT




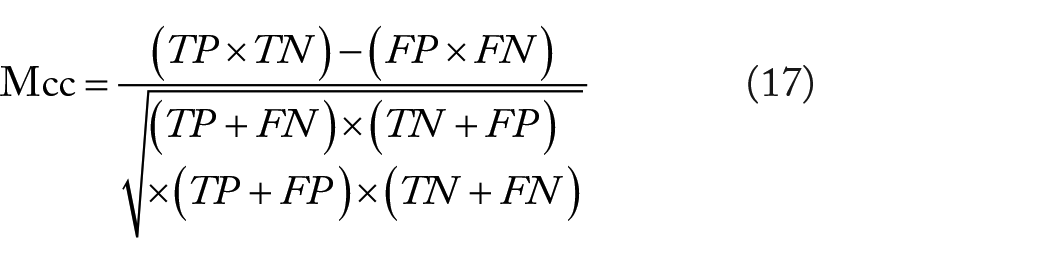
where Ac is the accuracy, Sn represents the sensitivity, Sp is the specificity, Pe represents the precision, and Mcc is Matthews’s correlation coefficient. TP and TN represent the number of true interacting and true noninteracting pairs that were correctly predicted, respectively. FP and FN are the count of true noninteracting pairs and true interacting pairs falsely predicted, respectively. In addition, we used receiver operating curve (ROC) to further evaluate the performance of RNN-SIFT in the experiment.
Results and Discussion
Performance of the proposed RNN-SIFT model
In the experiment, we used the yeast and human datasets to evaluate performance of the proposed RNN-SIFT model. Generally, overfitting will affect experimental results. Therefore, we divided the whole datasets into the training datasets and independent test datasets for preventing a biased evaluation. Specifically, we split the yeast dataset into 6 parts and selected 5 parts of them as the training set and the remaining dataset selected as independent test dataset. The human dataset was also processed by using the same strategy. Meanwhile, 5-fold cross-validation tests were also employed to evaluate prediction ability of the RNN-SIFT for fair comparison, and several parameters of the RNN model were optimized through using the grid search for ensuring fairness. Here, we set up the learning rate = 0.001, training step = 1000, and hidden units = 200. Tables 1 and 2 show the experimental results of the proposed RNN-SIFT model on the yeast and human datasets.
Fivefold cross-validation results shown using the RNN-SIFT model on yeast.

Abbreviations: Ac, accuracy; Mcc, Matthews’s correlation coefficient; Pe, precision; RNN, recurrent neural network; SIFT, scale invariant feature transform; Sn, sensitivity.
Fivefold cross-validation results shown using the RNN-SIFT model on human.

Abbreviations: Ac, accuracy; Mcc, Matthews’s correlation coefficient; Pe, precision; RNN, recurrent neural network; SIFT, scale invariant feature transform; Sn, sensitivity.
As can be seen from Table 1, the proposed RNN-SIFT model obtained good experimental results on yeast dataset. The result of average accuracy 94.34%, average sensitivity 67.12%, average precision 79.79%, and average Mcc 71.61% was achieved in the experiments on 5-fold cross-validation tests. Similarly, another promising finding from Table 2 was that the RNN-SIFT also achieved better prediction results on human dataset, whose average accuracy, sensitivity, precision, and Mcc are 97.12%, 83.70%, 85.24%, and 79.35%, respectively. As a result, the proposed RNN-SIFT model has high value in research.
The good experimental results for predicting SIPs are mainly attributed to use the SIFT feature extraction method and RNN classifier. The main advantage of the RNN-SIFT model is that the SIFT method can extract key evaluation features from PSSM, and the RNN classifier has the advantage of processing sequence data. As discussed, this is mainly due to the following 3 reasons: (1) PSSM contains not only the position information but also the evolution information of protein sequence and retains plenty of prior information. This makes it possible to contain a number of key features that can be extracted. (2) SIFT uses the concept of “scale space” to capture features at multiple scale levels, which not only increases the number of available features but also makes the method highly tolerant to scale changes. This makes it possible for extracting the evolutionary information embedded in PSSM and capturing SIP information. (3) Recurrent neural network has some characteristics in memory, parameter sharing, and Turing completeness, so which provide an advantage for learning based on the nonlinear characteristics of sequences. Therefore, RNN is used to perform classification for predicting SIPs. The results demonstrate 2 things. First, the SIFT method is very suitable for extracting SIP features. Second, the RNN classifier performs well for predicting SIPs, giving good results.
Comparison with the method of BPNN-based and SVM-based
It is interesting to note that the RNN-SIFT model is very suitable for predicting SIPs and can obtain good prediction results. However, to further evaluate the performance of the RNN-SIFT model, we compared the RNN classifier with the BPNN classifier and the SVM classifier by using the same SIFT approach on yeast and human datasets, respectively. To ensure fair comparison, several parameter settings of BPNN were optimized by employing grid search approach. Specifically, the epochs (the time of training), the eta (learning rate), the BS (the batch size of each training), and the WS (weights) of BPNN are set to 100, 0.006, 0.5, and 0.7. Similarly, by using the same strategy as described above, the RBF kernel parameters of the SVM were optimized, where c is 0.5 and g is 10.8 and other parameters should be take the default values. In addition, the SVM classifier used the LIBSVM tool 40 to perform classification.
Tables 3 to 6 below show the experimental results of BPNN-SIF and SVM-SIFT on the yeast and human datasets, respectively. Meanwhile, the comparison of ROC curves on the yeast and human datasets between RNN, BPNN, and SVM is shown in Figures 5 and 6 below, respectively. As outlined in Tables 3 and 4, the BPNN-SIFT model achieved 91.31% average accuracy and the SVM-SIFT model obtained 89.58% average accuracy on yeast dataset. Similarly, as can be seen from Tables 5 and 6, the results of average accuracy 93.84% and 91.79% are obtained by the BPNN-SIFT model and the SVM-SIFT model on human dataset, respectively. When comparing our results to those of BPNN-SIFT and SVM-SIFT, it must be pointed out that the performance of RNN classifier is significantly better than that of the other 2 classifiers. At the same time, from Figures 5 and 6, the ROC curves of RNN classifier are also significantly better than those of the other 2 classifiers. A major reason for good prediction results is that SIP sequence is nonlinear sequence data, and RNN classifier has some characteristics in memory, parameter sharing, and Turing completeness and can provide an advantage for learning based on the nonlinear characteristics of sequences. From the above analysis, we conclude that the proposed RNN-SIFT model is a useful tool for identifying SIPs, as well as other bioinformatics tasks.
Fivefold cross-validation results shown by using the BPNN-SIFT model on yeast.

Abbreviations: Ac, accuracy; BPNN, back propagation neural network; Mcc, Matthews’s correlation coefficient; Pe, precision; SIFT, scale invariant feature transform; Sn, sensitivity.
Fivefold cross-validation results shown by using the SVM-SIFT model on yeast.

Abbreviations: Ac, accuracy; Mcc, Matthews’s correlation coefficient; Pe, precision; SIFT, scale invariant feature transform; Sn, sensitivity; SVM, support vector machine.
Fivefold cross-validation results shown by using the BPNN-SIFT model on human.

Abbreviations: Ac, accuracy; BPNN, back propagation neural network; Mcc, Matthews’s correlation coefficient; Pe, precision; SIFT, scale invariant feature transform; Sn, sensitivity.
Fivefold cross-validation results shown by using the SVM-SIFT model on human.

Abbreviations: Ac, accuracy; Mcc, Matthews’s correlation coefficient; Pe, precision; SIFT, scale invariant feature transform; Sn, sensitivity; SVM, support vector machine.

Comparison of ROC curves between RNN, BPNN, and SVM on yeast dataset.

Comparison of ROC curves between RNN, BPNN, and SVM on human dataset.
Comparison with other methods
To go a step further and validate the performance of the proposed RNN-SIFT model, we compare the prediction results of the RNN-SIFT model with those of the previous methods, such as SLIPPER, 18 CRS, 31 SPAR, 31 DXECPPI, PPIevo, 41 and LocFuse. 42 Tables 7 and 8 show a detailed comparison results on the yeast and human datasets. It can be seen from Table 7 that the average accuracy of RNN-SIFT is obviously higher than that of the other 6 approaches on yeast dataset. Similarity, Table 8 displays the prediction accuracy obtained by the RNN-SIFT model is also significantly better than that of the other 6 methods on human dataset. A similar conclusion was reached by comparing the results from Tables 7 and 8 that the proposed RNN-SIFT model has an excellent prediction capability and can be used for predicting the quality of SIPs. This is a result of using a robust RNN classifier and an effectively SIFT feature extraction technique. These comparison results are further evidence that the RNN-SIFT is suitable for predicting SIPs.
Comparison results between RNN-SIFT and other methods on yeast dataset.
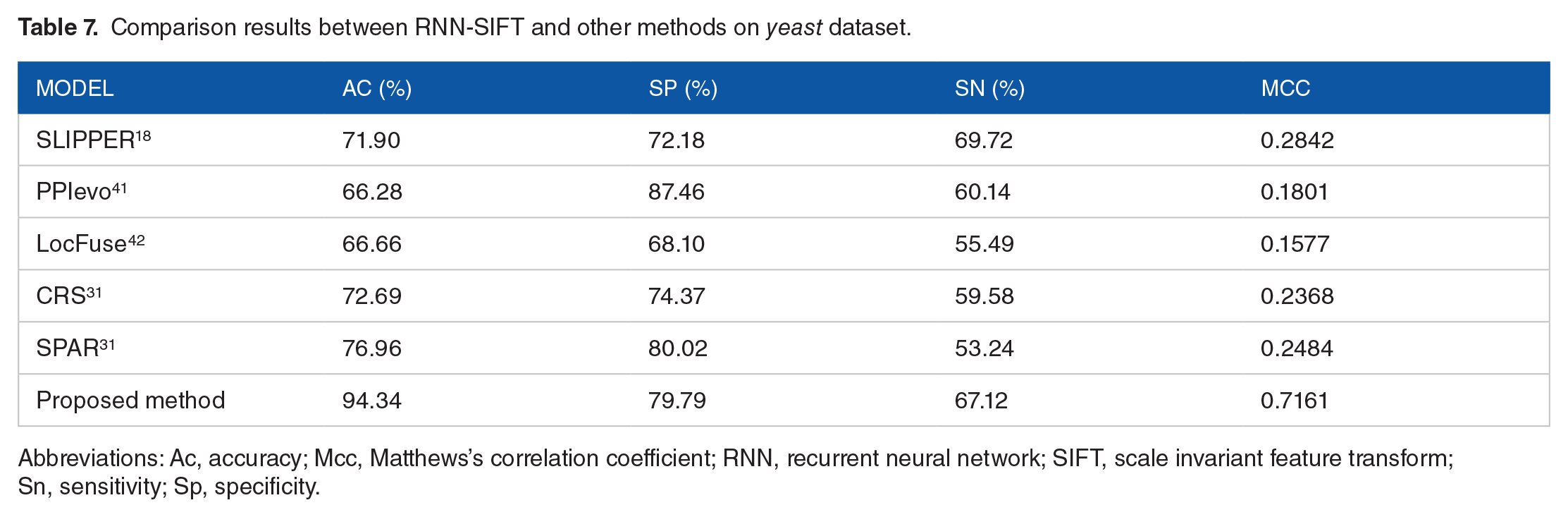
Abbreviations: Ac, accuracy; Mcc, Matthews’s correlation coefficient; RNN, recurrent neural network; SIFT, scale invariant feature transform; Sn, sensitivity; Sp, specificity.
Comparison results between RNN-SIFT and other methods on human dataset.
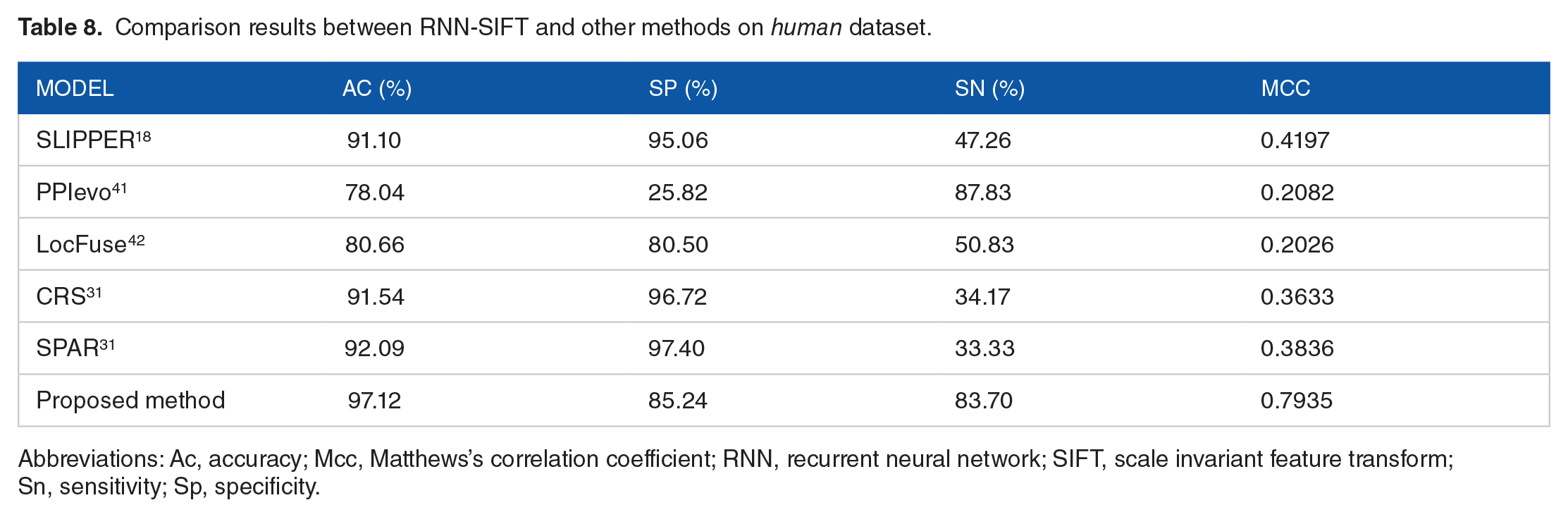
Abbreviations: Ac, accuracy; Mcc, Matthews’s correlation coefficient; RNN, recurrent neural network; SIFT, scale invariant feature transform; Sn, sensitivity; Sp, specificity.
Conclusions
In the study, we proposed a novel computational method called RRN-SIFT, which combines the RNN with SIFT for predicting SIPs based on protein evolutionary information. Extensive experiments show that the RRN-SIFT obtained an average accuracy of 94.34% and 97.12% on the yeast and human dataset, respectively. We also compared our performance with that of BPNN, the state-of-the-art SVM, and other exiting methods. By comparing with the experimental results, the performance of RNN-SIFT is significantly better than that of the BPNN, SVM, and other previous methods in the domain. This is mainly due to the following 3 reasons: (1) PSSM contains not only the position information but also the evolution information of protein sequence and retains plenty of prior information. This makes it possible to contain a number of key features that can be extracted. (2) Scale invariant feature transform uses the concept of “scale space” to capture features at multiple scale levels, which not only increases the number of available features but also makes the method highly tolerant to scale changes. This makes it possible for extracting the evolutionary information embedded in PSSM and capturing self-protein interaction information. (3) Self-interacting protein sequence is nonlinear sequence data, and RNN has some characteristics in memory, parameter sharing, and Turing completeness and can provide an advantage for learning based on the nonlinear characteristics of sequences. Therefore, we conclude that the proposed RNN-SIFT model is a useful tool for predicting SIPs, as well as to solve other bioinformatics tasks.
Footnotes
Acknowledgements
The authors would like to thank all the guest editors and anonymous reviewers for their constructive advices.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by “the Fundamental Research Funds for the Central Universities (2019XKQYMS88).”
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
J-YA conceived the algorithm, performed analyses, prepared the datasets, carried out experiments, wrote the manuscript, and approved the final manuscript.
Data Availability
In this study, our experimental datasets contain yeast and human dataset, which can be obtained from the publicly available DIP,23 BioGRID,24 IntAct,25 InnateDB,26 and MatrixDB.27